





Abstract
Phosphate binding plays an important role in modulating protein–protein interactions, which are ubiquitous in various biological processes. Accurate prediction of phosphate binding sites is an important but challenging task. Small size and diversity of phosphate binding sites lead to a substantial challenge for developing accurate prediction methods.
Here, we present the phosphate binding site predictor (PBSP), a novel and accurate approach to identifying phosphate binding sites from protein structures. PBSP combines an energy-based ligand-binding sites identification method with reverse focused docking using a phosphate probe. We show that PBSP outperforms not only general ligand binding sites predictors but also other existing phospholigand-specific binding sites predictors. It achieves ∼95% success rate for top 10 predicted sites with an average Matthews correlation coefficient value of 0.84 for successful predictions. PBSP can accurately predict phosphate binding modes, with average position error of 1.4 and 2.4 Å in bound and unbound datasets, respectively. Lastly, visual inspection of the predictions is conducted. Reasons for failed predictions are further analyzed and possible ways to improve the performance are provided. These results demonstrate a novel and accurate approach to phosphate binding sites identification in protein structures.
The software and benchmark datasets are freely available at http://web.pkusz.edu.cn/wu/PBSP/.
Supplementary data are available at Bioinformatics online.
1 Introduction
Protein–protein interactions play fundamental roles in various biological processes such as signal transduction, cell cycle regulation, metabolic regulation, immune response and gene expression regulation (Braun and Gingras, 2012; Humphrey et al., 2015; Keskin et al., 2016). Their spatial and temporal regulations are essential for the survival at the cellular and organism levels. Phosphorylation (Singh et al., 2017), one of the most pervasive and best-studied post-translational modifications (Gokirmak et al., 2010), can modulate the nature and the strength of protein–protein binding (Nishi et al., 2011). Indeed, adding or removing a dianionic phosphate group somewhere on a protein might change its physicochemical properties, folding stability, kinetics and dynamics (Johnson, 2009). Phosphorylation can also provide diverse and selective recognition sites for the binding of proteins containing phosphate-binding domains (Nishi et al., 2014). Numerous studies have identified and investigated various phosphate-binding domains, such as 14-3-3, WW, FHA, SH2, BRCT and WD40 domains (Jin and Pawson, 2012; Pawson et al., 2001; Reinhardt and Yaffe, 2013; Yaffe and Smerdon, 2001). However, the details of phosphate binding sites and recognition mechanisms are difficult to obtain because of difficulties and high costs in experiments. Thus, reliably predicting the region of phosphate binding sites can be useful in guiding mutagenesis experiments and protein functional annotation, and in the modeling of a phosphorylation-related protein complex structures.
In recent years, a variety of methods have been developed for binding sites prediction, such as MIB (Lin et al., 2016), ConCavity (Capra et al., 2009), MSPocket (Zhu and Pisabarro, 2011), LISE (Xie and Hwang, 2012), COACH (Yang et al., 2013), AutoSite (Ravindranath and Sanner, 2016), DeepSite (Jimenez et al., 2017) and P2Rank (Krivak and Hoksza, 2018). However, researches on the prediction of phosphate-binding sites are limited. Joughin et al. (2005) developed a method in which physical and chemical properties of nine phosphopeptide-binding domains were used to characterize the phosphopeptide-binding region on protein surface. Sanchez et al. (Ghersi and Sanchez, 2009; Hernandez et al., 2009) developed an energy-based approach called SiteHound, which determines molecular interaction fields for a protein structure using a phosphate oxygen probe in order to identify the potential binding sites for phosphorylated ligand. In fact, the two methods are developed to detect favorable binding regions instead of pinpointing the position in space of the phosphate group. Parca et al. (2011) developed Pfinder, a comparative method for the identification of binding sites for phosphate groups, both in the form of ions or as parts of other non-peptide ligands. This method relies heavily on template library, and it is unable to predict truly novel sites that have no analogues in template library. SiteHound performs much better than Pfinder in the identification of binding sites for phosphate group in both protein–protein interactions and protein-small molecule interactions, likely due to Pfinder relying on matching to existing phospholigand binding sites (Ghersi and Sanchez, 2012).
Small size and diversity of phosphate binding sites lead to a substantial challenge for developing accurate prediction methods. Most of the ligand-binding sites often locate among the largest pockets on protein surface (London et al., 2010; Nayal and Honig, 2006). However, the phosphate group is much smaller than general ligands, and in many instances the phosphate-binding sites may not locate in the largest pockets. The diversity of phosphate binding sites represent a second challenge to their prediction. Phosphate groups are ubiquitous in biology and nearly half of known proteins have been shown to interact with partners containing such a group (Hirsch et al., 2007). As phosphorylation occurs in such a diverse range of contexts, it is not surprising that the domains involved in its selective recognition are often unrelated from an evolutionary or structural standpoint. Although the residues preferentially bound to phosphate groups are somewhat conserved, in fact in many instances the binding site is not the region of most positive electrostatic potential on the protein surface (Ghersi and Sanchez, 2012). All in all, proteins interacting with phosphate are highly heterogeneous, and no single property can be used to reliably identify the phosphate binding sites.
Here, we present the phosphate-binding site predictor (PBSP), a method to predict phosphate binding sites given a protein 3D structure. PBSP combines a modified energy-based binding sites identification method with reverse focused docking to improve accuracy and selectivity. We show that PBSP outperforms other representative methods, and achieved ∼95% success rate for top 10 predicted sites with an average Matthews correlation coefficient values of 0.84 for successful predictions. Furthermore, PBSP can also provide phosphate binding modes at atomic resolution.
2 Materials and methods
2.1 Phosphate binding site definition
The phosphate binding site is defined as the spherical region of 7 Å surrounding the phosphorous atom of PO4 moiety in previous studies (Brakoulias and Jackson, 2004; Kinoshita et al., 1999). However, it yields larger binding sites than the actual ones, and some residues in the sites are not interacting with phosphate groups. In order to define the phosphate binding site more accurately, the distances between all heavy atoms in a receptor protein and phosphorus atom in its ligand were calculated across the whole Protein Data Bank (PDB) (Berman et al., 2000) (Supplementary Fig. S1a). The main peak in the normalized-distance distribution is mainly distributed within 4.7 Å. The P–O bond length in phosphate group is about 1.5–1.6 Å, thus most distances between the oxygen atoms in phosphate groups and the heavy atoms in receptors are within 3.2 Å, which is suitable for the formation of hydrogen bond. Therefore, receptor residue in which at least one heavy atom is within 4.7 Å from phosphorus atom is defined as phosphate-binding residue (Supplementary Fig. S1b). In order to make sure that phosphorylation plays important role in the interaction, one phosphate binding site should be composed of more than two phosphate binding residues.
2.2 Datasets
All the crystal structures in PDB (Berman et al., 2000) with resolution ≤3 Å and at least two chains were downloaded on July 06, 2020, and filtered for the presence of at least one phosphoresidue as indicated by the names ‘PTR’ (phosphotyrosine), ‘SEP’ (phosphoserine) or ‘TPO’ (phosphothreonine). Structures in which small molecules occur in the binding sites and affect the interactions were filtered out. Then the phosphate binding sites defined above were extracted from the crystal structures, and the chains containing phosphate binding sites were designated as phosphate binding proteins. Redundancy is removed by using PISCES (Wang and Dunbrack, 2003), a protein sequence culling server, with a sequence identity cutoff of 50%. It yielded a non-redundant bound dataset of 97 chains containing 106 phosphate binding sites. The detailed information of the phosphate binding sites and corresponding PDB entries is summarized (Supplementary Table S1). A corresponding dataset of unbound phosphate binding proteins was also generated by carrying out a BLAST (Camacho et al., 2009) search (default parameters) using the bound chain sequences against the entire PDB. Hits with sequence identity or coverage ≤ 98% were excluded. The structures with an empty binding sites and with the highest resolution were retained. Finally, this protocol yielded a non-redundant unbound dataset of 63 chains containing 67 phosphate binding sites (Supplementary Table S2).
2.3 PBSP
PBSP relies on and contains third-party software called AutoDockFR (Ravindranath et al., 2015) and AutoSite (Ravindranath and Sanner, 2016) with slight modification. As shown in Figure 1, the workflow of PBSP contains the following three steps:
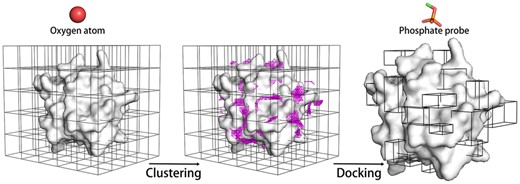
Flowchart of PBSP for phosphate binding site prediction
(i) Potential phosphate binding pockets identification. A few studies show that focused docking produce more accurate docking poses than blind docking (Ban et al., 2018; Ghersi and Sanchez, 2009; Liu et al., 2020). Thus, we first identify potential binding pockets for subsequent reverse focused docking. The first step requires the computation of a 1 Å resolution oxygen (AutoDock atom type OA, hydrogen bond acceptor) affinity map with AutoGrid4 (Huey et al., 2007) in AutoDock (Morris et al., 2009), using a box enclosing the entire protein. The map contains evenly spaced grids where each grid point yields the sum of the pairwise interaction energies between an oxygen-atom with all protein atoms. A predefined energy cutoff (-0.32 kcal/mol for all cases, Supplementary Fig. S2) is applied to filter out all the grid points with unfavorable interaction energies. Subsequently, the remaining grid points are clustered using a DBSCAN algorithm which was developed in AutoSite (Ester et al., 1996; Ravindranath and Sanner, 2016). The potential phosphate binding pocket is defined as the cluster generated by the clustering algorithm. In order to obtain more potential binding pockets, cVolcut which is a cutoff to filter small clusters in the clustering algorithm is set to 10 in PBSP.
(ii) Reverse focused docking. A phosphate probe (CH3-) was created and geometry optimization was carried out using Gaussian 16 (Baboul et al., 1999; Frisch et al., 2016; Grimme et al., 2010; Rassolov et al., 2001). For every potential phosphate binding pocket identified above, the probe is docked to a box encompassing the pocket using AutoDockFR (Ravindranath et al., 2015) with default parameters. Here, we developed two PBSP models, PBSP/R by rigid docking and PBSP/F by flexible docking. In PBSP/F, side chains of arginine, lysine, serine, threonine, histidine and tyrosine in the potential binding pocket are made flexible, and the docking box is large enough to allow side chains to rotate freely inside. This is because these six residues (Arg, Lys, Ser, Thr, His, Tyr) occur most favorably in phosphate binding sites (Supplementary Fig. S1).
In the case of PBSP/R, only the first two terms (i.e. EL-L or ligand intra-molecular and EL-RR or ligand-rigid receptor inter-molecular interactions) are considered. The additional terms (EL-FR, EFR-FR and EFR-RR) are automatically included in the scoring functions in PBSP/F. The EL-RR and EFR-RR terms are efficiently obtained by interpolating values in affinity maps. The remaining terms (EL-L, EL-FR, EFR-FR) are computed using explicit atom pairs for every non-bonded pair of atoms excluding 1-3 interactions, and 1-4 interactions not mediated by a rotatable bond (Ravindranath et al., 2015).
2.4 Evaluation
3 Results
3.1 Comparison with other methods
An energy-based phospholigand-specific binding-site predictor SiteHound (OP probe) (Hernandez et al., 2009) was chosen for comparison against PBSP, as well as four general ligand-binding sites prediction methods: a knowledge-based predictor LISE (Xie and Hwang, 2012), a geometry-based predictor MSpocket (Zhu and Pisabarro, 2011), a machine learning-based predictor P2Rank (Krivak and Hoksza, 2018) and a geometry-based predictor Fpocket (Le Guilloux et al., 2009). Figure 2 presents success rates of different methods for phosphate-binding site prediction in both bound and unbound datasets from top 1 to 10 predictions. A protein with at least one successful site (successful prediction) in top X (1 ≤ X ≤ 10) predictions is considered as a successful protein. And the success rate is defined as the proportion of the number of successful proteins to the total number of all proteins in the dataset. The two PBSP predictors have better performance than other methods, including SiteHound. For the bound dataset, PBSP/R and PBSP/F achieve 89.6% and 85.6% success rate in top 5 predictions, and 93.4% and 95.3% success rate in top 10 predictions, respectively. While SiteHound makes 72.6% and 86.6% success rate in top 5 and 10 predictions, respectively. And for the unbound dataset, PBSP/R and PBSP/F achieve 85.1% and 79.1% success rate in top 5 predictions, and 95.5% and 95.5% success rate in top 10 predictions, respectively. While SiteHound makes 67.2% and 86.6% success rate in top 5 and 10 predictions, respectively. The average values of MCC, Sen, Spe, Acc and Prec of successful sites in top 10 predictions of successful proteins in the dataset are given in Table 1. The results show that, for both PBSP/R and PBSP/F methods and in both bound and unbound datasets, MCC, Specificity, Accuracy and Precision of PBSP are higher than those of other methods. Only Sensitivity of PBSP is slightly lower than that of SiteHound. That is because SiteHound identifies large putative binding sites, while PBSP identify small sites with high precision. As shown in Table 2, the putative sites predicted by SiteHound are about three times larger than that of crystal structure and PBSP. SiteHound uses a phosphate oxygen probe to identify favorable energy grid points, and then grid points are clustered to form the putative binding sites. While PBSP uses oxygen atom to identify potential phosphate binding pockets, and then a phosphate probe is docked to the pocket to generate an optimal conformation from which phosphate binding site is extracted. For a range of applications such as guiding biochemical experiments, it is important to keep the predicted site as small as possible without compromising accuracy (Laurie and Jackson, 2005).
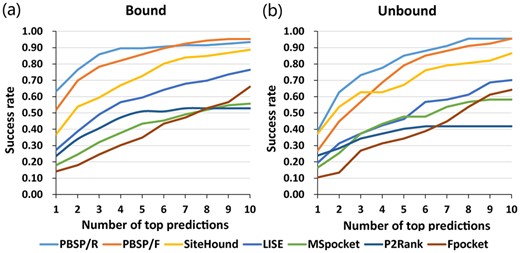
Success rate comparison of different methods for phosphate binding site prediction in both bound and unbound dataset from top 1 to 10 prediction. A protein with at least one successful site (successful prediction) in top X (1 ≤ X ≤ 10) predictions is considered as a successful protein. And the success rate is defined as the proportion of the number of successful proteins to the total number of all proteins in the dataset
Performance comparison of different methods on both bound and unbound datasets
| Method | Bound | Unbound | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCC | Sen | Spe | Acc | Prec | MCC | Sen | Spe | Acc | Prec | |
| PBSP/R | 0.920 | 0.913 | 0.999 | 0.997 | 0.937 | 0.779 | 0.760 | 0.996 | 0.991 | 0.822 |
| PBSP/F | 0.868 | 0.872 | 0.997 | 0.994 | 0.878 | 0.794 | 0.792 | 0.996 | 0.991 | 0.821 |
| SiteHound | 0.559 | 0.988 | 0.948 | 0.949 | 0.343 | 0.556 | 0.966 | 0.949 | 0.950 | 0.346 |
| LISE | 0.644 | 0.700 | 0.989 | 0.983 | 0.631 | 0.661 | 0.731 | 0.988 | 0.981 | 0.636 |
| MSpocket | 0.467 | 0.904 | 0.921 | 0.921 | 0.281 | 0.460 | 0.886 | 0.930 | 0.929 | 0.279 |
| Fpocket | 0.619 | 0.883 | 0.97 | 0.968 | 0.468 | 0.566 | 0.844 | 0.966 | 0.963 | 0.416 |
| P2Rank | 0.586 | 0.858 | 0.968 | 0.966 | 0.443 | 0.549 | 0.814 | 0.962 | 0.958 | 0.431 |
| Method | Bound | Unbound | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCC | Sen | Spe | Acc | Prec | MCC | Sen | Spe | Acc | Prec | |
| PBSP/R | 0.920 | 0.913 | 0.999 | 0.997 | 0.937 | 0.779 | 0.760 | 0.996 | 0.991 | 0.822 |
| PBSP/F | 0.868 | 0.872 | 0.997 | 0.994 | 0.878 | 0.794 | 0.792 | 0.996 | 0.991 | 0.821 |
| SiteHound | 0.559 | 0.988 | 0.948 | 0.949 | 0.343 | 0.556 | 0.966 | 0.949 | 0.950 | 0.346 |
| LISE | 0.644 | 0.700 | 0.989 | 0.983 | 0.631 | 0.661 | 0.731 | 0.988 | 0.981 | 0.636 |
| MSpocket | 0.467 | 0.904 | 0.921 | 0.921 | 0.281 | 0.460 | 0.886 | 0.930 | 0.929 | 0.279 |
| Fpocket | 0.619 | 0.883 | 0.97 | 0.968 | 0.468 | 0.566 | 0.844 | 0.966 | 0.963 | 0.416 |
| P2Rank | 0.586 | 0.858 | 0.968 | 0.966 | 0.443 | 0.549 | 0.814 | 0.962 | 0.958 | 0.431 |
Note: The values are the average values of the successful sites in top 10 predictions of successful proteins in the dataset. Bold values denote the best performance in each category.
Performance comparison of different methods on both bound and unbound datasets
| Method | Bound | Unbound | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCC | Sen | Spe | Acc | Prec | MCC | Sen | Spe | Acc | Prec | |
| PBSP/R | 0.920 | 0.913 | 0.999 | 0.997 | 0.937 | 0.779 | 0.760 | 0.996 | 0.991 | 0.822 |
| PBSP/F | 0.868 | 0.872 | 0.997 | 0.994 | 0.878 | 0.794 | 0.792 | 0.996 | 0.991 | 0.821 |
| SiteHound | 0.559 | 0.988 | 0.948 | 0.949 | 0.343 | 0.556 | 0.966 | 0.949 | 0.950 | 0.346 |
| LISE | 0.644 | 0.700 | 0.989 | 0.983 | 0.631 | 0.661 | 0.731 | 0.988 | 0.981 | 0.636 |
| MSpocket | 0.467 | 0.904 | 0.921 | 0.921 | 0.281 | 0.460 | 0.886 | 0.930 | 0.929 | 0.279 |
| Fpocket | 0.619 | 0.883 | 0.97 | 0.968 | 0.468 | 0.566 | 0.844 | 0.966 | 0.963 | 0.416 |
| P2Rank | 0.586 | 0.858 | 0.968 | 0.966 | 0.443 | 0.549 | 0.814 | 0.962 | 0.958 | 0.431 |
| Method | Bound | Unbound | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCC | Sen | Spe | Acc | Prec | MCC | Sen | Spe | Acc | Prec | |
| PBSP/R | 0.920 | 0.913 | 0.999 | 0.997 | 0.937 | 0.779 | 0.760 | 0.996 | 0.991 | 0.822 |
| PBSP/F | 0.868 | 0.872 | 0.997 | 0.994 | 0.878 | 0.794 | 0.792 | 0.996 | 0.991 | 0.821 |
| SiteHound | 0.559 | 0.988 | 0.948 | 0.949 | 0.343 | 0.556 | 0.966 | 0.949 | 0.950 | 0.346 |
| LISE | 0.644 | 0.700 | 0.989 | 0.983 | 0.631 | 0.661 | 0.731 | 0.988 | 0.981 | 0.636 |
| MSpocket | 0.467 | 0.904 | 0.921 | 0.921 | 0.281 | 0.460 | 0.886 | 0.930 | 0.929 | 0.279 |
| Fpocket | 0.619 | 0.883 | 0.97 | 0.968 | 0.468 | 0.566 | 0.844 | 0.966 | 0.963 | 0.416 |
| P2Rank | 0.586 | 0.858 | 0.968 | 0.966 | 0.443 | 0.549 | 0.814 | 0.962 | 0.958 | 0.431 |
Note: The values are the average values of the successful sites in top 10 predictions of successful proteins in the dataset. Bold values denote the best performance in each category.
Average number of residues in successful predicted binding sites
| Method | In crystal structure | PBSP/R | PBSP/F | SiteHound | LISEa | MSpocket | Fpocketb | P2Rank |
|---|---|---|---|---|---|---|---|---|
| Number | 4.3 | 4.1 | 4.2 | 13.0 | 4.7 | 17.9 | 5.1 | 10.4 |
| Method | In crystal structure | PBSP/R | PBSP/F | SiteHound | LISEa | MSpocket | Fpocketb | P2Rank |
|---|---|---|---|---|---|---|---|---|
| Number | 4.3 | 4.1 | 4.2 | 13.0 | 4.7 | 17.9 | 5.1 | 10.4 |
Lise outputs the centers of the predicted site, from which the residues within a sphere of default 5.5 Å are extracted and considered as putative binding residues.
972 sets of parameters of Fpocket were tested, and performance of the best set of parameters is shown.
Average number of residues in successful predicted binding sites
| Method | In crystal structure | PBSP/R | PBSP/F | SiteHound | LISEa | MSpocket | Fpocketb | P2Rank |
|---|---|---|---|---|---|---|---|---|
| Number | 4.3 | 4.1 | 4.2 | 13.0 | 4.7 | 17.9 | 5.1 | 10.4 |
| Method | In crystal structure | PBSP/R | PBSP/F | SiteHound | LISEa | MSpocket | Fpocketb | P2Rank |
|---|---|---|---|---|---|---|---|---|
| Number | 4.3 | 4.1 | 4.2 | 13.0 | 4.7 | 17.9 | 5.1 | 10.4 |
Lise outputs the centers of the predicted site, from which the residues within a sphere of default 5.5 Å are extracted and considered as putative binding residues.
972 sets of parameters of Fpocket were tested, and performance of the best set of parameters is shown.
Among the above methods, only PBSP uses molecular docking. To assess the importance of docking, we compare the prediction results of PBSP and AutoSite in both clustering stage and docking stage. In fact, PBSP clustering stage is a variation of the AutoSite algorithm. AutoSite uses three atom types (carbon, oxygen and hydrogen) with a cVolcut of 50 to identify binding sites, while PBSP only uses oxygen atom with a cVolcut of 10. The comparison betwen AutoSite (cVolcut = 50), AutoSite (cVolcut = 10), PBSP clustering stage, AutoSite (cVolcut = 50) + CH3PO42- rigid docking, AutoSite (cVolcut = 10) + CH3PO42- rigid docking, PBSP/R and PBSP/F is shown in Supplementary Figure S3. AutoSite (cVolcut = 10) + CH3- rigid docking is comparable with PBSP/R in bound dataset, but slightly worse in unbound dataset. AutoSite (cVolcut = 50) and AutoSite (cVolcut = 10) are also slightly worse than PBSP clustering stage. The average numbers of potential binding pockets per protein identified by AutoSite (cVolcut = 50), AutoSite (cVolcut = 10) and PBSP clustering stage are shown in Supplementary Tables S3 and S4. PBSP clustering stage produces more potential binding pockets than AutoSite. From those results, we can conclude three points. First, CH3- docking is very important in identifying phosphate binding sites, which can be used to improve the performances of other methods. Second, it is necessary to provide enough potential binding pockets to ensure that the correct site is in them. Third, the choice of atom type in predicting the binding sites also affects the results. In fact, docking also has been used to predict ligand-binding site in other studies (Fukunishi and Nakamura, 2011; Heo et al., 2014; Hetenyi and van der Spoel, 2011; Wu et al., 2018). However, those methods mainly focus on small molecule binding, and the whole ligand is docked to the receptor, which are not suitable for protein–protein binding systems because of the complexity of protein–protein docking. Here, we assume the phosphate group contribute significantly to the binding of the whole phosphopeptide or phosphoprotein, and used a small phosphate probe instead of the whole phospholigand to identify potential binding sites by reverse focused docking, which significantly reduces the amount of calculation.
Compared with other methods, PBSP is relatively time-consuming because of the use of docking (Supplementary Table S5). However, PBSP performs much better than other methods, and the prediction speed is acceptable in practical application. In this article, the default number of evaluations of the scoring function is used in PBSP docking stage. So most dockings fail to converge (Supplementary Table S6), but it doesn't affect the performance of PBSP (Supplementary Fig. S4).
3.2 Phosphate binding modes prediction
PBSP not only predicts the phosphate binding sites precisely but also provides structural information of the binding modes. As shown in Figure 3, we calculated the distributions of distances between the position of phosphate atom in crystal structures and that in predicted phosphate binding modes, for all successful cases. The distances are mainly distributed within 4 Å, and the average of distances are 1.4 and 2.4 Å in bound and unbound dataset, respectively. Compared with Pfinder, in which the average of distances are 5 Å in holo dataset (Parca et al., 2011), PBSP is more reliable and accurate. This may be due to the use of reverse focused docking, which is originally used for identifying potential protein targets for a small-compound ligand (Xu et al., 2018). However, because of the use of docking, PBSP is more sensitive to the conformational changes of protein structures than other methods. In Table 1, other methods give similar performances (eg. MCC) for bound and unbound datasets, while PBSP gives much better MCC for bound dataset. For binding modes predictions (Fig. 3), the performance of PBSP in unbound dataset is also worse than that in bound dataset. Nevertheless, when considering top 10 predictions for each case, the successful rate from unbound dataset can reach ∼95%, as high as that from bound dataset. Therefore, our PBSP can be well applied in real applications when the complex structure is unknown, although the true binding mode may not be captured in the top-1 prediction.
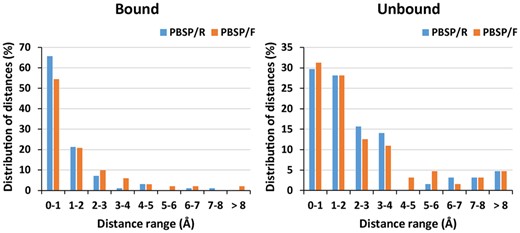
Distribution of distances between the position of phosphate atom in crystal structures and the corresponding predicted positions for all successful cases in both bound and unbound datasets
We use flexible docking in PBSP/F, to try to improve the performance on unbound form structures. And it works well in some cases. In bound-state structure of human p56lck SH2 domain (Tong et al., 1996), both PBSP/R and PBSP/F can predict the position of phosphate accurately (Fig. 4a, b). In the unbound-state structure, the conformation of the loop containing phosphate-binding residues E157 and S158 are located changes greatly, which make it hard to predict the site. Compared with PBSP/R, PBSP/F utilizing flexible docking improves the prediction performance of the unbound form of this protein (Fig. 4c, d).
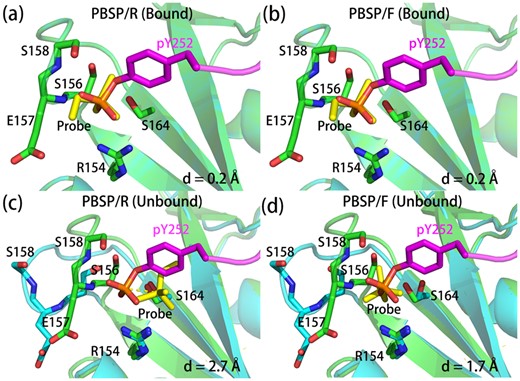
Comparison of predicted phosphate binding modes and experimentally determined structures. The phosphate binding protein in bound form crystal structure (1lkk) and that in predicted binding modes are shown in green and cyan, respectively. And phosphopeptides in bound form crystal structures and phosphate probes in predicted binding modes are shown in magenta and yellow, respectively. The distances between phosphate atom in bound form crystal structure and the phosphate atom in predicted binding mode are indicated as d. For (a) and (b), the bound form structure was used to predict the binding mode by PBSP, and for (c) and (d), the unbound form structures (1bhh) was used
Although PBSP/F improves the performance in some cases, overall PBSP/F performs slightly worse than PBSP/R. The reason lies in the differences in the rankings of phosphate binding sites between PBSP/R and PBSP/F. There are about 53 putative phosphate binding pockets and only one true phosphate binding pocket per protein in PBSP (Supplementary Table S3 and S4). If the phosphate probe finds a more comfortable pose in the putative pockets than in the true pocket, the true pocket will be ranked behind the putative pockets. This is more likely to happen in PBSP/F than in PBSP/R because flexible docking improves the pose of the phosphate probe not only in the true pocket but also in other putative pockets. In PBSP/F, the phosphate probe is in a more comfortable pose in the pockets, and the corresponding docking scores of pockets are lower than that in PBSP/R.
3.3 Analysis of unsuccessful cases
As listed in Supplementary Table S7, unsuccessful cases are different between PBSP/R and PBSP/F predictions. Further analysis by visual inspection of the unsuccessful cases is conducted, and these cases can be classified into four different categories:
(i) The phosphate binding pocket has been identified as the one in crystal structure, but not the precise binding residues. For example, protein tyrosine phosphatase 10D (Madan and Gopal, 2011), whose prediction result is shown in Figure 5a. Phosphate binding residues in crystal structure are K154, A244 and Q286, while K154, C242, S243 and R248 are predicted by PBSP/R. The phosphate atom has been shifted by only 3.3 Å comparing crystal structure and the predicted binding mode. Interestingly, in crystal structure, the interaction between the phosphate group and the receptor protein is mediated by two water molecules. However, the effect of water molecules is not taken into account in PBSP.
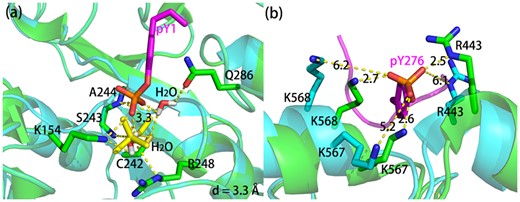
(a) The bound form structure (3s3h) in green was used to predict phosphate binding site by PBSP/R, and phosphopeptide in bound form structures and phosphate probe in predicted binding mode are shown in magenta and yellow, respectively. (b) The bound form (4ch2) and the unbound form (4nzq) structure are shown in green and cyan, respectively. The phosphopeptide in bound form structure is shown in magenta
(ii) The true phosphate binding site does not rank in the top 10 predictions. There are two possible reasons, inaccuracy of scoring function and too many putative phosphate binding sites. It is assumed that the true phosphate binding site would exhibit stronger affinity to the phosphate probe than the other sites. However, PBSP uses a semiempirical free energy force field to score the predictions, which make it hard to accurately calculate the binding affinities. Conservative information can be added into PBSP to improve the performance of scoring function as other method did (Ghersi and Sanchez, 2012). Another reason may be that there are too many putative phosphate binding sites with the potential to bind phosphate. However, which one bind to phosphate in crystal structure depends on not only the binding of phosphate but also the other part of the phospholigand. Based on this work, our further research will focused on predicting the binding of entire phosphopeptide.
(iii) Conformational changes during phosphate binding. Ligand binding can involve a wide range of induced conformational changes in proteins, such as loop or domain movements. In some cases, PBSP correctly predicts the phosphate binding sites in the bound form structure, but not the unbound form structure. As shown in Figure 5b, the conformation of the unbound form structure of thrombin (Lechtenberg et al., 2014) changes greatly, which make it hard for PBSP/R to predict the phosphate binding sites. Although flexible side-chain docking is used in this article to try to improve the performance and it works well in some specific cases, overall the performance in unbound dataset is not improved. Since the beginning of the field of docking, conformational changes in proteins induced by binding have confounded protein docking algorithms by greatly increasing the degrees of freedom to be sampled (Marze et al., 2018). While rotamer libraries have alleviated the sampling challenges for surface sidechains (Krivov et al., 2009), backbone flexibility remains the principal challenge in protein docking. In order to tackle backbone flexibility, molecular dynamics, Monte Carlo approaches and even machine learning have steadily advanced toward reliably capturing large conformational changes in protein docking (Harmalkar and Gray, 2021).
(iv) A small phosphate binding site. Some small phosphate binding sites can be missed in the energy-based phosphate binding pockets identification in PBSP, because they are too small to contribute significant interaction energies. In order to solve this problem, different docking box identification methods can be combined to generate enough docking boxes on the protein surface to encompass the missed small phosphate binding sites.
4 Conclusion
In this article, we present a novel and accurate approach for predicting phosphate binding sites in phosphorylation-dependent protein–protein interactions: PBSP. Firstly, candidate ligand-binding sites are identified from a calculated oxygen-atom affinity map using a predefined energy cutoff and DBSCAN clustering analysis. The results are then used to guide reverse focused dockings of a phosphate probe, to obtain predictions with improved accuracy and selectivity. PBSP not only can identify phosphate binding sites more accurate and precise than other methods but also can provided structural information of phosphate binding modes. Average distances between the phosphate atoms in crystal structures and that in predicted binding modes are 1.4 and 2.4 Å in bound and unbound dataset, respectively. Analysis of unsuccessful cases implies that there are some aspects that affect the performance of PBSP, such as conformational changes of unbound form structure, the ranking of the putative phosphate binding sites and the binding free energy contribution from other parts of the phospholigand. In conclusion, PBSP performs much better than other methods, which suggests that it can be useful in guiding mutagenesis experiments, protein functional annotation, and in the modeling of a phosphorylation-related protein complex structures.
Acknowledgements
The authors thank Shenzhen Bay Laboratory Supercomputing Center for providing computational resources. They are very grateful to the authors of the software ADFRsuite for their excellent work and freely available software. They thank Dr. Zhi-Qiang Ye for helpful suggestions.
Funding
The authors were grateful to the Key-Area Research and Development Program of Guangdong Province [2020B0101350001], the National Natural Science Foundation of China [21933004] and the Shenzhen Fundamental Research Program [GXWD20201231165807007-20200812124825001] for financial support.
Conflict of Interest: none declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}