





Abstract
Bisulfite sequencing (BS-seq) is currently the gold standard for measuring genome-wide DNA methylation profiles at single-nucleotide resolution. Most analyses focus on mean CpG methylation and ignore methylation states on the same DNA fragments [DNA methylation haplotypes (mHaps)]. Here, we propose mHap, a simple DNA mHap format for storing DNA BS-seq data. This format reduces the size of a BAM file by 40- to 140-fold while retaining complete read-level CpG methylation information. It is also compatible with the Tabix tool for fast and random access. We implemented a command-line tool, mHapTools, for converting BAM/SAM files from existing platforms to mHap files as well as post-processing DNA methylation data in mHap format. With this tool, we processed all publicly available human reduced representation bisulfite sequencing data and provided these data as a comprehensive mHap database.
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is an epigenetic mark that plays an important role in gene regulation, development and tumorigenesis (Greenberg and Bourc'his, 2019). Traditional array-based approaches are limited to measuring mean DNA methylation of individual CpG sites and neglect the within-sample heterogeneity of the profiled cell populations. Sequencing-based techniques such as whole genome bisulfite sequencing (WGBS) and reduced representation bisulfite sequencing (RRBS) are now widely used to measure DNA methylation at single-nucleotide resolution, which also enables researchers to characterize DNA methylation patterns at the sequencing read-level. Even when generated from bulk data, DNA methylation data of a single read fragment is guaranteed to stem from a single chromosome of a single cell. Thus, the methylation pattern of CpGs on each fragment represents a discrete DNA methylation haplotype (mHap) (Shoemaker et al., 2010). Motivated by this concept, Landau et al. (2014) presented the concept of the Proportion of Discordant Reads (PDR) to quantify within-sample tumor heterogeneity, which is one of the most widely used read-level summary statistics (Teschendorff and Relton, 2018). Similarly, Guo et al. (2017) proposed the metric of Methylation Haplotype Load (MHL) to quantify the level of coordinated methylation. Scherer et al. (2020) systematically benchmarked six types of heterogeneity scores including PDR, MHL, entropy, epipolymorphism and two newly proposed metrics. Recently, Xu et al. (2021) introduced a Cell Heterogeneity-Adjusted cLonal Methylation (CHALM) score which uses DNA mHap patterns to quantify the functional consequences of DNA methylation. However, the calculation of mHap-level summary statistics requires raw sequence alignments, which are usually not publicly available due to privacy issues and large file sizes.
To overcome these shortcomings, we propose mHap, a novel file format for DNA methylation bisulfite sequencing (BS-seq) data. It is designed to interface between DNA mHaps and downstream analyses, including extraction of per-base methylation metrics, identification of differentially methylated regions, characterization of discordant and concordant methylation patterns. We also developed a C++ software called mHapTools for converting raw BS-seq alignments (SAM/BAM) generated by popular aligners, such as BSMAP (Xi and Li, 2009), Bismark (Krueger and Andrews, 2011) and BS-seeker (Chen et al., 2010), to mHap files. Finally, we collected 3731 publicly available RRBS raw sequencing datasets and corresponding annotation information to establish a comprehensive mHap database.
2 mHap format and mHapTools description
The mHap format file was developed for efficient storage and manipulation of DNA mHaps. It is a genomic position-based format with six fields, including the genomic region specified by the first and last CpG sites in the first three fields, the DNA mHap and the numbers of reads with given mHaps and strands (Fig. 1A). In the fourth field, DNA mHaps are represented as binary strings of 0 and 1, where 1 indicates methylated CpG site and 0 indicates unmethylated CpG site. An mHap file can be indexed by Tabix (Li, 2011) to achieve random and fast retrieval of haplotypes overlapping with a specified chromosomal region. To demonstrate the features of the mHap format, we compared it to other formats utilized by existing BS-seq programs (Supplementary Table S1). For example, BSMAP (Xi and Li, 2009), MethylQA (Sun et al., 2013), BRAT (Harris et al., 2010), BRAT-BW (Harris et al., 2012) and METHPIPE (Song et al., 2013) generate standard BAM/SAM files as output. Bismark (Krueger and Andrews, 2011), BS-Seeker (Chen et al., 2010) and BS-Seeker2 (Guo et al., 2013) output BAM files with additional tags to represent methylation states for CpG and non-CpG sites (Supplementary Fig. S1). However, BAM files contain raw sequencing reads and can be used to determine genetic information which is an important source of individual’s privacy. In most public data repositories such as GEO (Gene Expression Omnibus) and EGA (European Genome-phenome Archive), BAM files are treated as raw data and protected by default without explicit consent from participants. It is thus widely accepted by the research community that summary data such as mean methylation is a good balance between privacy conserving and data sharing. Other related tools such as Bismark methylation extractor (Krueger and Andrews, 2011), MethylDackel (Ryan, 2017), CGmapTools (Guo et al., 2018) and METHCOMP (Peng et al., 2018) output methylation summaries free of sequence information, including bedGraph, CGmap and bedMethyl. However, these formats only report aggregated methylation signal at each cytosine site, and neglect read-level methylation information. In contrast, mHap is a general DNA mHap format and retains complete read-level CpG methylation.
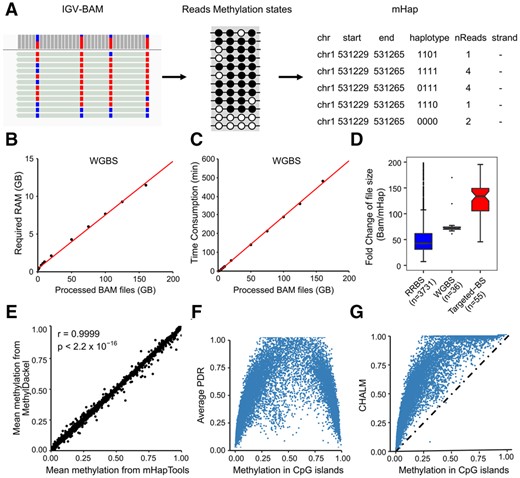
The DNA mHap format. (A) Conversion of a BAM file to the mHap format file. The left panel shows the IGV view of a typical RRBS BAM file; the middle panel displays the methylation states in each read, in which black and white circles represent methylated and unmethylated CpG cytosines, respectively; and the right panel shows the mHap file format. (B and C) RAM and time consumptions in the processing of WGBS data. BAM files of different sizes were subsampled from a high coverage WGBS sample SRX175348. (D) Compression ratios of mHap format for different types of BS-seq data. (E) Consistency in CpG island-level mean methylation obtained by mHapTools and MethylDackel, using a CLL sample (SRX885188) as an example. (F and G) Distributions of PDR and CHALM for human CpG islands against mean methylation. PDR, CHALM and mean methylation levels were calculated for 23 228 CpG islands covered by 10 or more reads with at least 4 CpG sites in each read
To manipulate DNA mHaps in the mHap format, we developed mHapTools that can parse alignments of SAM/BAM format files from different platforms, and convert them to mHap files. The mHapTools is fast and memory efficient for large-size BS-seq data. Especially, the maximum amount of RAM and running time are both linearly correlated with BAM file sizes (Fig. 1B and C and Supplementary Fig. S2). For a typical RRBS BAM file, the processing time is ∼10 min for a personal computer with 500 MB of RAM (Supplementary Fig. S2A and C). For a high coverage WGBS BAM file of 150 GB, it finishes in 8 h with the maximum memory usage of ∼10 GB (Fig. 1B and C). More importantly, the mHap format dramatically reduces the size of a BAM file (40- to 140-fold) while retaining complete read-level CpG methylation information (Fig. 1D).
To explore how mHap format achieve this significant file size reduction, we tested the effects of different procedures in converting BAM files to mHap files including information reduction, aggregation and gzip compression (Supplementary Table S2 and Fig. S3). In information reduction step, DNA mHaps were extracted from BAM files (Supplementary Fig. S1C). Surprisingly, this step only results in 2-fold file size reduction for RRBS, 4- and 1.3-fold for WGBS and targeted BS-Seq, respectively. In the aggregation step, reads with the same haplotypes were merged and the count column was updated to reflect number of reads with the same haplotypes. This step results another 3.5-fold reduction in file size for RRBS, 2.5-fold for WGBS and 20.7-fold for targeted BS-Seq. In particular, samples with higher coverage benefit more from this step. The last Gzip compression step introduces additional 6-fold reduction for all three assays. In summary, three steps together result in 39-fold reduction in file size for RRBS, 69-fold for WGBS and 148-fold for targeted BS-Seq (Fig. 1D). We also observed that additional binarization only results in minimal changes in file size and thus not adopted by current version of mHapTools (Supplementary Fig. S4).
To validate the implementation of mHapTools, we next compared mean methylations of all annotated CpG islands derived from MethylDackel (Ryan, 2017) and mHapTools. Using a chronic lymphocytic leukemia (CLL) sample as an example, we observed consistent results by these two methods (Fig. 1E). Using the same data, we also tested two read-level measurements, PDR and CHALM, and showed their relationships with mean methylation (Fig. 1F and G), which are also in accordance with previous results (Landau et al., 2014; Xu et al., 2021). However, our calculation is based on the space-saving mHap format files, the processing time and memory usage are drastically reduced. All functions are described on mHapTools websites with vignettes illustrating the basic and advanced features.
3 Applications and data curation
Locally disordered methylation is deemed a hallmark of cancer and its association with gene expression was extensively characterized in CLL (Landau et al., 2014). This type of analysis requires access to DNA mHaps which were usually not available unless raw data were shared. Our mHap format coupled with mHapTools represents a framework to store, share and analyze BS-seq data in haplotype-level (Supplementary Fig. S9). In our previous study, we have demonstrated that extraembryonic ectoderm (ExE) and cancer share similar DNA methylation landscapes (Smith et al., 2017). We here conducted a comprehensive analysis between the epiblasts and ExE cells. Genes with significantly differences in promoter PDR, but not in mean methylation, show strong enrichment in many developmental pathways that are repressed in ExE and activated in Epiblasts (Supplementary Figs S5–S8). These genes might otherwise be missed using traditional mean methylation-based methods. We further characterized cancer-specific discordant methylation using CCLE dataset (Ghandi et al., 2019). Interestingly, a significant proportion of these promoters have no reliable changes in mean methylation, especially for AML and kidney cancers (Supplementary Figs S10–S12).
To demonstrate the usage of mHapTools, we processed 3731 human RRBS samples for over 60 tissues across 57 diseases types with a unified pipeline (Supplementary Methods). Sample annotations, quality control reports, mean methylation as well as mHap files were organized as a database that is freely accessible for academic use (http://mhap.sciplus.cloud). The next version of this database will expand significantly to include all publicly available human WGBS samples. Note that this database is different from the ASMdb (Zhou et al., 2020), which aims to identify DNA mHap regions with allele-specific DNA methylation.
4 Conclusion
Here we propose mHap, a novel file format for DNA mHaps. It is lightweight and efficient to store read-level CpG methylation information obtained using various BS platforms. The mHap file is extremely compact in size and supports fast retrieval of mHaps in specified regions. We also developed mHapTools for manipulating mHap files, such as converting BAM/SAM files to mHap files, merging multiple mHap files, and extracting useful information from mHap files. In the future, more haplotype-level summary statistics such as those quantifying comethylation will be developed. With our tool, we created a comprehensive DNA mHap database without storing genomic information. The mHap format, together with mHapTools and mHap database, serve as the building blocks for the analysis of DNA mHaps.
Acknowledgements
The mHap database was developed by Shanghai Technology Development Co., Ltd. We are grateful for the high performance computing center of the Center for Excellence in Molecular Cell Science (CEMCS), CAS, for its support in data processing.
Funding
This study is supported by the Hundred Talents Program of the Chinese Academy of Sciences, the Shanghai Pujiang Program [20PJ1414700 to J.S.] and National Natural Science Foundation of China [61972257 to X.Z.].
Conflict of Interest: none declared.
References
Author notes
Zhiqiang Zhang and Yuhao Dan authors wish it to be known that these authors contributed equally.


{kind=link}