





Abstract
Processing raw reads of RNA-sequencing (RNA-seq) data, no matter public or newly sequenced data, involves a lot of specialized tools and technical configurations that are often unfamiliar and time-consuming to learn for non-bioinformatics researchers. Here, we develop the R package BP4RNAseq, which integrates the state-of-art tools from both alignment-based and alignment-free quantification workflows. The BP4RNAseq package is a highly automated tool using an optimized pipeline to improve the sensitivity and accuracy of RNA-seq analyses. It can take only two non-technical parameters and output six formatted gene expression quantification at gene and transcript levels. The package applies to both retrospective and newly generated bulk RNA-seq data analyses and is also applicable for single-cell RNA-seq analyses. It, therefore, greatly facilitates the application of RNA-seq.
The BP4RNAseq package for R and its documentation are freely available at https://github.com/sunshanwen/BP4RNAseq.
Supplementary data are available at Bioinformatics online.
1 Introduction
The assessment of gene expression is central to uncovering the functions of the genome, understanding the regulation of development and investigating the molecular mechanisms that underlie cancer and other diseases. Various methods thus have been developed to quantify the transcriptome. As the advancements of the next-generation sequencing technology, RNA-sequencing (RNA-seq) now becomes the routine to assess the genome-wide gene expression due to its high speed, accuracy and reproducibility and low cost (Marioni et al., 2008). Thousands of RNA-seq studies have been conducted to address key biological questions as mentioned above (Ma et al., 2020; Qi et al., 2020; Stark et al., 2019; Xie et al., 2020). Besides conducting a brand new RNA-seq study, retrospectively analyzing an enormous volume of RNA-seq data that have been accumulating and deposited in public data repositories, such as the Gene Expression Omnibus (GEO, 128224 datasets; Edgar, 2002) and the Sequence Read Archive (SRA, 1869440 datasets; Leinonen et al., 2011), can be a crucial exploratory analysis for the hypothesis development (Grace et al., 2018) and experiment design (Hart et al., 2013), or an important new biological study using solely public data or combining them with newly generated data from samples (Rung and Brazma, 2013). However, despite its power and the allures that it brings, processing RNA-seq data and translating them from raw reads into a meaningful quantified gene expression matrix, although usually not the research interest, involve a lot of specialized algorithms and software. Choosing the right tools, such as quality control, alignment and assembly tools, and corresponding parameter settings and connecting these tools correctly and smoothly need specialized knowledge and sometimes programming skills. These are normally absent and time-consuming to learn for researchers without bioinformatic background and thus impede the potential applications of RNA-seq.
Some efforts have been devoted to facilitate the use of public RNA-seq data by compiling and summarizing raw data into transcript and gene counts ready for downstream analyses (Supplementary Table S1). The lag between the availability of the latest raw RNA-seq data in public repositories and the processing and inclusion of them into compiled datasets motivates the development of the software that can help users to download and convert public RNA-seq data into counts (Supplementary Table S1). Despite their usefulness, however, the complied datasets and software still involve a lot of queries and technical configurations that are time-consuming to do or grasp. Further, they do few favors when researchers additionally need to process their own sequenced data. Moreover, a recent study suggests that combining alignment-based and alignment-free transcript quantification, which are the two main contrasting RNA-seq analysis workflows now existing, may achieve better performance than using one of them alone (Lachmann et al., 2020). Most of compiled datasets and software available, unfortunately, only implement either workflow (Supplementary Table S1). This, on one hand, may decrease the sensitivity and accuracy of RNA-seq analysis in terms of the number of detected expressed genes and the assessment of gene expression levels and on the other hand, may hamper the comparison or combination of RNA-seq datasets that are processed by different workflows.
To address these limitations of previous tools and promote the applications of RNA-seq among researchers, we develop an R package—BP4RNAseq. The package integrates the state-of-the-art bioinformatics tools, implements an optimized pipeline and serves as a ‘babysitter’ for retrospective and newly generated RNA-seq data analyses. Users can just provide two non-technical parameters, such as data accession id in the public repositories and the species name (scientific or common name) or species name and the type of reads, i.e. single-end reads or paired-end reads, then the package will translate remote or local raw RNA-seq reads into formatted gene expression quantification with high sensitivity and accuracy at both gene and transcript levels ready for downstream analyses based on both alignment-based and alignment-free transcript quantification protocols.
2 Implementation and performances
The BP4RNAseq is developed using R language. Its implementation is based on an optimized pipeline integrated with the state-of-the-art tools from downloading the RNA-seq data and the reference genome, transcript and corresponding annotation files from public repositories to counting the expressed genes and transcripts (Fig. 1). Specifically, first, public RNA-seq data are downloaded with Entrez Direct and converted to FASTQ format with SRA Toolkit. This step is unique to the retrospective analysis of public RNA-seq data. Then, reads quality and the existence of the adapter are checked with FASTQC (Andrews, 2019) and reads are trimmed, if necessary, with Cutadapt (Martin, 2011). Subsequently, the reference genome, transcript and corresponding annotation files are downloaded using NCBI Datasets. Finally, gene expression is assessed using HISAT2 (Kim et al., 2019) and StringTie (Pertea et al., 2015) for alignment-based quantification and Salmon (Patro et al., 2017) for alignment-free quantification. Several studies have shown that these two pipelines have better performances in their respective workflows (Lachmann et al., 2020; Sahraeian et al., 2017).
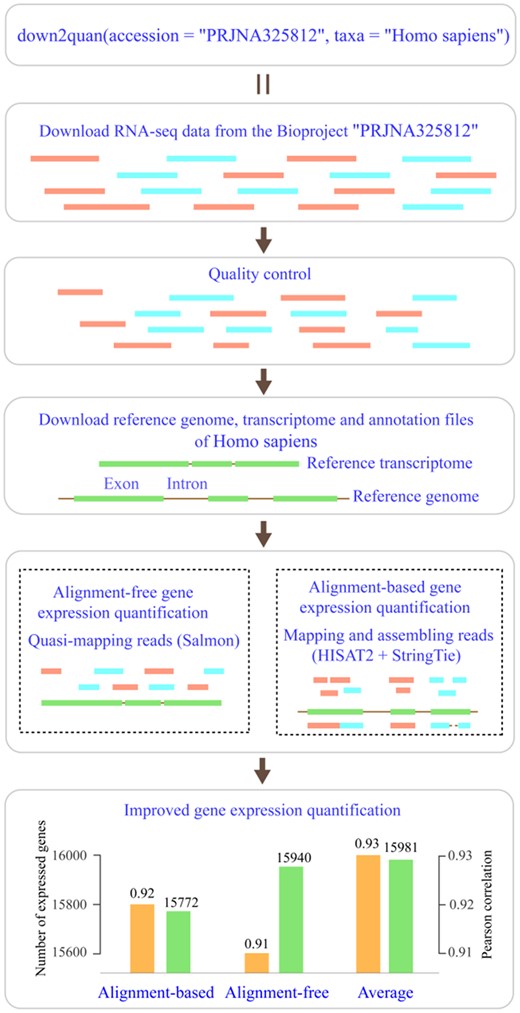
The workflow of BP4RNAseq. A simple one-line code ‘down2quan(accession = “PRJNA325812”, taxa = “Homo sapiens”)’ with the default settings is performed. First, the tool automatically downloads all the RNA-seq data from the Bioproject ‘PRJNA325812’ with a size of 44.3 GB after converted to FASTQ files and does the quality control. Then, it downloads 3.79 GB reference genome, transcript and annotation files and performs the reads alignment and gene expression quantification. Outputs from the tool are gene expression quantification based on alignment-based workflow and alignment-free workflow. Evaluation of the consistency between RNA-Seq derived fold changes and qPCR derived fold changes of the differential expression of 18 080 protein-coding genes shows that averages over the two workflows detected more expressed genes and had higher Pearson correlation to qPCR derived fold changes (bottom subfigure; for details about the evaluation, see main text). To work with their interested public data, researchers only need to modify the ‘accession’ and the ‘taxa’ parameters with no more physical and mental efforts. For analyzing local RNA-seq data in FASTQ format, users can use the fastq2quan() tool
All these tools with their corresponding optimized parameters are integrated into two highly automated main functions, i.e. down2quan and fastq2quan, that take two non-technical parameters. The down2quan function can be used to process public data with the data (the whole bioproject, biosample/biosamples or a single dataset) accession id in the NCBI repositories and the species name (scientific or common name), while fastq2quan is used to process researchers’ own data with species name and the type of reads, i.e. single-end reads or paired-end reads. Outputs from both functions are two gene count matrixes and two transcript count matrixes based on the alignment-based workflow and the alignment-free workflow, and corresponding average matrixes over two workflows. Although the package focuses on bulk RNA-seq analyses, it can also be extended to process single-cell RNA-seq data, whether remote or local data, based on the Alevin algorithm integrated with the Salmon (Srivastava et al., 2019; details about single-cell RNA-seq analysis are given in the github page and package vignette). Moreover, the package supports the parallel computing.
To evaluate the performance of default settings of our package and the sensitivity and accuracy of gene expression averages over alignment-based workflow and alignment-free workflow, we reanalyzed RNA-seq data (GSE83402) from a previous study, where the expression of 18 080 protein-coding genes was compared between RNA-seq assessments and qPCR assessments (Everaert et al., 2017). RNA-seq data were quantified with the default settings using the reference genome and corresponding annotation files downloaded from Ensembl to facilitate the comparison with qPCR results. The differential expression analyses of RNA-seq data were performed using DESeq2 (Love et al., 2014). Low expressed genes with a total count less than 10 were prefiltered based on the official recommendation of DESeq2 to reduce the potential biases. qPCR between 11 and 32 were used in the analyses following the protocol in Everaert et al. (2017) to evaluate the consistency between RNA-Seq derived fold changes and qPCR derived fold changes. We found that the overall alignment rate of the RNA-seq data was 77.7%, which is slightly higher than 77.2% reported in Everaert et al. (2017). In total, 15 981 of 18 080 genes were detected using the average matrix, and lower numbers were found using alignment-based workflow matrix and alignment-free workflow matrix, i.e. 15 772 and 15 940, respectively (Fig. 1). All matrixes identified more genes than the 13 045 genes detected in Everaert et al. (2017). Moreover, all the matrixes showed high Pearson correlations to the qPCR derived fold changes, while the average matrix had the highest concordance to qPCR results (Fig. 1). Overall, these results support the robustness of default settings of our package and suggest that the average matrix over two workflows can improve the sensitivity and accuracy of RNA-seq analyses. Additionally, we offer full control to experts to fine tune and optimize individual steps in the analyses, such as the quality control step and the reads alignment step, using individual tools integrated in our package (more details can be found in the package help page and vignette). This also allows users to inspect the intermediate outputs and may further improve the performance of RNA-seq analyses. Researchers may use the averages for downstream analyses. Alternatively, we recommend to decide the type of matrixes to use based on their consistencies with qPCR results if available or/and the results from the downstream analyses.
3 Conclusion
RNA-seq is an indispensable tool in molecular biology but could also be applied in other areas such as ecology (Pascoal et al., 2018), plant science (Nagano et al., 2019), etc. However, the technical obstacles in the way of converting raw reads into a formatted gene expression quantification matrix have hurdled its applications. The BP4RNAseq aims to provide a ‘babysitter’ tool to minimize efforts from researchers and to improve the sensitivity and accuracy of RNA-seq analyses. It is a highly automated tool and receives only two non-technical parameters by taking care of all the technical details for the researchers. It is different from another sets of tools, such as Rsubread which lacks downloading (RNA-seq data and reference genome, transcript and corresponding annotation files) and quality control functions (Liao et al., 2019) and easyRNASeq which receives only inputs of trimmed and aligned reads (Delhomme et al., 2012). Additionally, we provide a bash script that can install all the dependencies including R. The R environment, which BP4RNAseq is based on, is rich in downstream analysis and visualization tools, such as edgeR (Robinson et al., 2010), DESeq2 (Love et al., 2014) and limma (Ritchie et al., 2015) for bulk RNA-seq analyses, and LTMG (Wan et al., 2019) and IRIS-EDA (Monier et al., 2019) specialized for single-cell RNA-seq analyses, and thus can facilitate the subsequent analyses which most researchers are interested in.
Acknowledgement
The authors thank three anonymous reviewers whose comments improved the quality and clarity of this article.
Funding
The work was supported by the National Key R&D Program of China [2018YFC0910405] and the National Natural Science Foundation of China [61922020, 61771165, 91935302].
Conflict of Interest: none declared.


{kind=link}