





Abstract
Motivation: Counting the number of occurrences of every k-mer (substring of length k) in a long string is a central subproblem in many applications, including genome assembly, error correction of sequencing reads, fast multiple sequence alignment and repeat detection. Recently, the deep sequence coverage generated by next-generation sequencing technologies has caused the amount of sequence to be processed during a genome project to grow rapidly, and has rendered current k-mer counting tools too slow and memory intensive. At the same time, large multicore computers have become commonplace in research facilities allowing for a new parallel computational paradigm.
Results: We propose a new k-mer counting algorithm and associated implementation, called Jellyfish, which is fast and memory efficient. It is based on a multithreaded, lock-free hash table optimized for counting k-mers up to 31 bases in length. Due to their flexibility, suffix arrays have been the data structure of choice for solving many string problems. For the task of k-mer counting, important in many biological applications, Jellyfish offers a much faster and more memory-efficient solution.
Availability: The Jellyfish software is written in C++ and is GPL licensed. It is available for download at http://www.cbcb.umd.edu/software/jellyfish.
Contact: gmarcais@umd.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Given a string S, we are often interested in counting the number of occurrences in S of every substring of length k. These length-k substrings are called k-mers and the problem of determining the number of their occurrences is called k-mer counting.
Counting the k-mers in a DNA sequence is an important step in many applications. For example, genome assemblers using the overlap-layout-consensus paradigm, such as the Celera (Miller et al., 2008; Myers et al., 2000) and Arachne (Jaffe et al., 2003) assemblers, use k-mers shared by reads as seeds to find overlaps. Statistics on the number of occurrences of each k-mer are first computed and used to filter out which k-mers are used as seeds. Such k-mer count statistics are also used to estimate the genome size: if a large fraction of k-mers occur c times, we can estimate the sequencing coverage to be approximately c and derive an estimate of the genome size from c and the total length of the reads. In addition, in most short-read assembly projects, errors are corrected in the sequencing reads to improve the quality of the final assembly. For example, Kelley et al. (2010) use k-mer frequencies to assess the likelihood that a misalignment between reads is a sequencing error or a genuine difference in sequence. A third application is the detection of repeated sequences, such as transposons, which play an important biological role. De novo repeat annotation techniques find candidate regions based on k-mer frequencies (Campagna et al., 2005; Healy et al., 2003; Kurtz et al., 2008; Lefebvre et al., 2003). The counts of k-mers are also used to seed fast multiple sequence alignment (Edgar, 2004). Finally, k-mer distributions can produce new biological insights directly. Sindi et al. (2008) used k-mers frequencies with large k (20 ≤ k ≤ 100) to study the mechanisms of sequence duplication in genomes.
We consider the k-mer counting problem in the context where the input string S is either one DNA sequence or a concatenation of many DNA sequences, and the alphabet is Σ = {A, C, G, T}. The main application to which we apply our new k-mer counting algorithms here is counting k-mers in sequencing reads from large genome sequencing projects where the length n of the sequence to process is equal to the length g of the genome sequenced times the coverage c of the sequencing project (n = g · c). Recent sequencing techniques, using shorter reads with a much deeper coverage (Schatz et al., 2010), generate large amounts of sequence and provide with a major challenge for genome assembly and for k-mer counting. For example, the giant Panda (Li et al., 2010) sequencing project generated 73× coverage yielding 176 GB of sequence, much larger than the 5 − 10× coverage a sequencing project using traditional Sanger methodology would generate.
Of course, k-mer counting can be naively implemented using a simple hash table, where keys are the k-mers and the stored values are the counts. However, this strategy is extremely slow and implementing multithreaded access to the hash table via standard locking mechanisms results in slower performance than a single-threaded implementation (Michael and Scott, 1996; Purcell and Harris, 2005). Typically, more advanced k-mer counters such as Tallymer (Kurtz et al., 2008) have been based on the suffix array data structure. Despite the recent algorithmic progress to compute the suffix array of a string, it remains a relatively expensive computational operation. Moreover, in sequencing applications, memory requirements for a suffix array grow linearly with the product of the coverage and genome size. Meryl, the k-mer counter built into the Celera assembler, uses a sorting-based approach that sorts the k-mers in lexicographical order; however, sorting billions of records quickly with limited memory is a challenging problem.
In order to process this huge increase in amount of sequence, the increasing availability of parallelism must be exploited. While the raw power of each processing core has leveled off, the number of cores per CPU is rising. Shared memory machines with 16 or more cores and 4 GB or more memory per core are commonly available in research facilities. Creation of parallel algorithms taking advantage of such a large number of cores in a shared memory environment is both a challenge and an opportunity: on the one hand, current programming paradigms either do not take advantage of the parallelism available or are difficult to implement; on the other hand, very fast programs using fine grain parallelism can be implemented.
In recent years, the MapReduce programing paradigm (Dean and Ghemawat, 2008) has been used to harness the computational power of large clusters of machines. The problem of counting k-mers is easy to implement on top of a MapReduce cluster, but the straightforward implementation, where the map operation emits all k-mers associated with a 1, incurs a large overhead. To reduce this overhead, one needs to increase the amount of work done on each node at the map stage by, for example, using a fast single-machine k-mer counter like Jellyfish on each node. The map operation then emits pairs of k-mers and their counts on a subset of the data. In that sense, the use of the MapReduce paradigm and optimized k-mer counters like Jellyfish are orthogonal.
Other processing architectures such as GPU computing have also been recently exploited for achieving faster parallel execution. However, the widespread availability of multi-core CPUs make them the first and easiest choice to program, and this is likely to remain true for some time to come. CPU development is not staying idle, and facilities, such as the CAS operation (Section 3.2) and the SSE extension (Section 3.4), are available in all modern CPUs to help achieve greater parallel execution.
Our k-mer counting algorithm is designed for shared memory parallel computers with more than one core. It uses several lock-free data structures that exploit a widely available hardware operation called ‘compare-and-swap’ (CAS) to implement efficient shared access to the data structures. In particular, Jellyfish uses lock-free queues (Ladan-mozes and Shavit, 2004; Michael and Scott, 1996) for communication between worker threads and a lock-free hash table (Michael, 2002; Purcell and Harris, 2005) to store the k-mer occurrences counts (see Section 3.2). Unlike a traditional data structure where access by multiple threads must be serialized by the use of a lock, lock-free data structures can be used concurrently by many threads while still preserving a coherent internal state. The lock-free data structure also provides better performance by increasing the amount of time spent in parallel execution versus serial execution. The efficient parallel data structures allow Jellyfish to count k-mers much faster than existing k-mer counting software. In our testing on a large assembly project (Section 4.1), Jellyfish takes minutes instead of hours.
Jellyfish is also very memory efficient. It implements a key compression scheme that allows it to use a constant amount of memory per key in the hash table for most applications, regardless of the length k of the k-mers counted (see Section 3.4). It also uses a bit-packed data structure to reduce wasted memory due to memory alignment requirements (see Section 3.3). In addition, unlike with suffix arrays, the expected storage requirement for the hash table does not grow linearly as the coverage increases. This property makes hash tables extremely attractive for use in the context of short-read sequencing projects. Jellyfish can be more than twice as memory efficient as other programs (Section 4.1).
Our results show that Jellyfish can count k-mers an order of magnitude or more faster than existing programs (Section 4.1). This suggests that lock-free hash tables are valuable for k-mer counting and possibly also in other problems where large strings must be processed. In addition, Jellyfish's novel memory-efficient key compression approach (Section 3.4) allows the hash table to use a similar amount of storage as suffix arrays in most common uses. Jellyfish has been adopted by the Quake error corrector (Kelley et al., 2010) and will be included in version 6.2 of the Celera Assembler (Miller et al., 2008). Our implementation of the lock-free hash table is general and may be of use in other applications.
2 METHODS
2.1 Execution profiling
All testing and timing was performed on a 64 bit x86 AMD Opteron machine with 32 cores at 2.5 GHz and 256 GB of RAM running Linux kernel version 2.6.31. The disks are RAID-10 with sustained write throughput of 260 MB/s. The time is the wall clock time measured with the GNU time utility averaged over five runs (except runs that exceed 1 h which are run only once).
To measure the memory usage, the programs were run under strace which logs every system call made by a process and its threads. The logs of the strace were parsed to compute the amount of memory used by the process by looking for the following system calls, which are the only calls available to request memory from the kernel on a Linux system: brk, mmap, munmap and mremap. In addition, the script counts only the memory areas that are writable for the process. Read-only pages are not counted as most of them correspond to the shared libraries. In some cases, this undercounts the true memory usage. For example, Tallymer maps the entire input sequence into read-only memory and accesses it in a random fashion, so all these read-only pages need to be present in memory. Jellyfish also maps the entire input sequence into read-only memory, but the sequence is accessed in a sequential fashion and only the current page needs to be present in memory. Memory usage and running time are measured in different runs as the strace mechanism can affect the running time of IO heavy programs. The overall CPU and IO load are measured with the Linux vmstat utility.
2.2 Sequence datasets
The M.gallopavo reads were taken from the Turkey genome assembly project (Dalloul et al., 2010). These short reads, from Roche 454 and Illumina GAII technology, total ∼24 GB of sequence for a genome of 1 GB. To vary coverage, we took a random sample of reads to obtain the desired amount of sequence.
The sequence of Homo sapiens (3 GB), Drosophila ananassae (3.5 GB of reads, genome of 189 MB), Coxiella burnetii (35.6 GB of reads, genome of 1.99 MB) and Zea mays (33 GB of reads, genome of 2 GB) were downloaded from the NCBI. The human sequence is the reference genome hs_ref_GRCh37 and, unlike all other datasets, consists of assembled chromosomes instead of sequencing reads.
2.3 Comparing with existing k-mer counters
We used two versions of Meryl (5.4 and 6.1) from the k-mer package of the Celera assembler (Miller et al., 2008), with all default options and 32 threads. To work around an issue with Meryl version 5.4 when dealing with a multifasta file, all the reads where concatenated with a single N as a separator. The original multifasta file was used with Meryl version 6.1.
Tallymer comes from version 1.3.4 of the genometools package (Kurtz et al., 2008). It was run as shown in the example of Tallymer's documentation given in the distribution. The Tallymer subroutine suffixerator used options -dna -pl -tis -suf -lcp, and the subroutine tallymer mkindex used options -minocc 1 -maxocc 10000000000 so that all k-mer counts would be written to disk.
3 ALGORITHM
3.1 A fast k-mer hash table
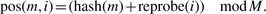
In our implementation, we maintain the length n of the hash table to be a power of two, M = 2ℓ for some ℓ, and the key representing the k-mer is encoded as an integer in the set Uk = [0, 4k − 1]. The function hash is a function mapping Uk into [0, M − 1]. The design of a function hash is described in Section 3.4.
When a new mer is added to the hash table, we attempt to store it in pos(m, 0), and if that position is already filled with a different key, we try pos(m, 1) and so on up to some limit. Here, we use a quadratic reprobing function: reprobe(i) = i(i + 1)/2. This reprobing function has a good behavior with respect to the usage of the hash table (Cormen et al., 1990) while not growing too fast, which is important for quickly sorting the hash table elements when writing the results to disk (see Section 3.5).
This straightforward standard scheme is both extremely slow when parallelized in the typical way using locks, and memory inefficient. In order to make it practical, we implement a lock-free strategy for allowing parallel insertions of keys and updates to values. We also design an encoding scheme to limit the storage used for both key and its associated counts. These are described in the following sections.
3.2 Updating the lock-free hash table
A lock, such as POSIX's pthread_mutex, can serialize access to the hash table and permits its use in a multi-threaded environment. However, if such a lock is used no concurrency is achieved, and therefore there is no gain in speed in the updates of the hash table. In addition, the overhead of maintaining the lock is incurred.
To allow concurrent update operations on the hash, we implement a lock-free hash table with open addressing (Purcell and Harris, 2005). Such lock-free hash tables exploit the CAS assembly instruction that is present in all modern multi-core CPUs. The CAS instruction updates the value at a memory location provided that the memory location has not been modified by another thread. Technically (see Algorithm 1), a CAS operation does the following three operations in an atomic fashion with respect to all of the threads: reads a memory location, compare the read value to the second parameter of the CAS instruction and if the two are equal, write the memory location with the third parameter of the CAS instruction. If two threads attempt to modify the same memory location at the same time, the CAS operation can fail. The CAS operation returns the value previously held at the memory location. Hence, one can determine if the CAS operation succeeded by checking that the returned value is equal to the old value. Unlike a lock that serializes the access to some shared resource, the CAS operation only detects simultaneous access to a shared memory location. It is then the responsibility of the calling thread to take appropriate action in the event that a conflict has been detected.

The main operation (Algorithm 2) supported by the hash is to increment the value associated with a key without using any locks. The value increment algorithm works in two steps. First it finds the location in the hash table that already holds the key or it claims an empty slot to store the key if the key is not present in the hash table. Second, it increments the value associated with the key.
Lines 1–7 in Algorithm 2 accomplish the first step. It finds an appropriate slot using the hash function and then does a CAS operation assuming that the entry in the hash is empty. If the returned value of the CAS operation is either EMPTY or equal to key, then that position is used for storing the key. Otherwise, there is a key collision: the reprobe value is incremented and we start over. The procedure fails if the maximum number of reprobes has been reached. Lines 8–12 accomplish the second step: they increment the value in an atomic way again using the CAS operation.

Two assumptions particular to k-mer counting simplify the design of the hash table. First, no entry is ever deleted and there is no need to maintain special information about deleted keys, such as tombstones (Purcell and Harris, 2005). Second, for k-mer counting the required size of the hash table should be easy to estimate or potentially the entire available memory is used. Hence, in the event that the hash table is full, it will be written to disk instead of doubling its size in memory (Gao et al., 2004; Shalev and Shavit, 2006). See Section 3.5 for more details.
3.3 Reduced memory usage for a hash entry
Our implementation uses a bit-packed data structure, i.e. entries in the hash table are packed tightly instead of being aligned with computer words. Albeit more complex to implement, especially in concert with the word-aligned CAS operation, and incurring a small computational cost, such bit-packed design is much more memory efficient and makes further memory saving schemes (variable length field and key encoding) worthwhile.
In addition, using a value field large enough to encode the number of occurrences of the most highly repeated k-mer is a waste of memory. Typically, with a deep coverage sequencing of a genome and a sufficiently large k, the majority of k-mers appear only once, as they are unique due to sequencing errors and because most genomic sequences are not composed of repeats. Most of the remaining k-mers occur approximately c times, where c is the sequencing coverage. A small number of k-mers, depending on the repetitiveness of the genome, occur a large number of times. To account for this, Jellyfish uses a small value field and allows a key to have more than one entry in the hash table: key, v1 and key, v2. The value associated with this key is then the number obtained by the concatenation of the bits v1v2. Moreover, to avoid the repetition of the key in the second entry, we only store a pointer (encoded as a number of reprobes) back to the previous entry. The now unused bits in the key field are used by the value field.
3.4 Space-efficient encoding of keys
The fact that an entry occurs at a known position in the hash table can be exploited to compactly store keys in the hash table in order to save a significant amount of additional memory. We choose a function f : Uk → Uk that is a bijection for which we can easily compute both f and its inverse, and set hash(m) = f(m) mod M. The length M = 2ℓ of the hash table is a power of 2, and the modulo M operation in the definition of the hash and pos functions (Equation 1) merely selects the ℓ lower bits of the sum f(m) + reprobe(i). Hence, provided the value of reprobe(i) is known, the position of a (key, value) pair in the hash table already encodes for the lower ℓ bits of f(m). Therefore, we store the 2k − ℓ higher bits of f(m) concatenated with bits representing the reprobe count i + 1 in the key field of the hash. We use i + 1 rather than i since 0 is reserved to indicate the entries that are still empty (EMPTY in Algorithm 2).
Conversely, given this content of the key field at position x, it is easy to find the sequence of the corresponding k-mer that is stored at this position. The key field contains the 2k − ℓ high bits of f(m) and the number of reprobes i. The lower ℓ bits of the f(m) can be therefore be retrieved by computing x − reprobe(i) modM. Finally, the k-mer m can be recovered by computing the inverse of f.
This scheme requires little modification to Algorithm 2. In particular, the keys do not need to be computed using the procedure described in the previous paragraph in order to be tested for equality (line 7). This is because if the content to be stored in the key field at a given position x for two k-mers m1 and m2 are equal, then by definition the reprobe value for both k-mers, the 2k − ℓ higher bits of f(mi) and their position in the hash are equal, thus f(m1) = f(m2) and m1 = m2 by the assumption that f is a bijection.
For the bijective f function, we use f(m) = A · m, where A is a 2k × 2k invertible matrix on ℤ/2ℤ. Here, m and A · m are interpreted either as integers or as 2k binary vectors.
Let ℋ = {x ↦ A · x mod 2ℓ ∣ A is invertible} be the set of all hash functions. This set is almost an universal set of hash functions (Cormen et al., 1990) in the following sense: the size of ℋ and the number N of matrices A for which A · x ≡ A · y (mod 2ℓ) satisfy N ≈ |ℋ|/2ℓ, provided that 22k ≫ 1 and 22k−ℓ ≫ 1 (see Appendix Section A.1 in Supplementary Material). In other words, the definition of the universal set of hash functions is satisfied within a small error, and the property of having few expected collisions is preserved by this approximation. The cases where this approximation breaks are the ‘easy’ cases corresponding to a small number of possible k-mers (22k close to 1) or a hash table big enough to contain almost all the k-mers (22k−ℓ close to 1).
The matrix A is chosen by iteratively drawing uniformly a random matrix out of the 24k2 possible binary matrices of this size, until it is not singular. This process terminates after an expected four iterations regardless of the size 2k (see Appendix A.1 in Supplementary Material). Faster algorithms to find an invertible matrix exist (Randall, 1991), but would have no impact on the execution speed of Jellyfish. Thus, an invertible, bijective hash function that is efficiently computable and that reduces the storage per key significantly is achieved.
To compute the binary matrix product A · m, we use the Streaming SIMD Extensions (SSE) instruction set of modern processors, if available. SSE instructions work on large registers (128 bits), treating them as vectors (e.g. of four 32 bit integers or two 64 bit integers). An SSE instruction performs the same operation on each element of the vector (or on each pair of elements of a pair of vectors) in parallel. For a 44 × 44 binary matrix A required to hash 22-mers, the SSE implementations computes 34.5 million multiplications per second on our test system versus 19.4 million multiplications per second for the C++ implementation that does not use SSE.
Surprisingly, in many applications, the above scheme uses an amount of space per key that is independent of the length of the k-mer and the length of the input string. Often k is chosen so that the event that a given k-mer appears more than once in the input sequence of size n is significant. For example, in sequencing reads, where n = c · g is the coverage times the genome length, such a k permits discrimination between k-mers really coming from the genome, which occur more than once, and error k-mers, which occur only once with high probability.
So k is chosen large enough so that the expected number of occurrences of a k-mer in a random string of length n is 1. In this case, the length k and the size of the hash table M = 2ℓ are such that the size of the key field |key|, which contains the 2k − ℓ high bits of f(key) and the reprobe count, is independent of k and n.
Suppose there are n k-mers in the input chosen at random, then each has an expected number of occurrences of μk = 4−kn = 1. Hence k = ⌈log4(n)⌉. Under these conditions, the marginal additional key space cost per extra nucleotide (Δk/Δn = 1/(n log 4)) decreases to 0 as n → ∞.
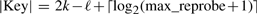
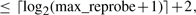
3.5 Fast merging of intermediate hash tables
Once computed, the hash table is written to disk as a list of (key, value) records. The list is sorted according to the lower l bits of the hash value of the mers, which is pos(m, 0), and ties are broken lexicographically. Sorting the output has the advantage that the results can be queried quickly using a binary search. More interestingly, it has the advantage that it allows two or more hash tables to be merged into one easily. This situation occurs when there is not enough memory to carry out the entire computation and intermediary results are saved to disk. Jellyfish will detect when the hash table needs to expand beyond the available memory and will instead write the current k-mer counts to disk, clear the hash table and begin counting afresh. The intermediate results can be merged in limited memory as described below.
In memory, the entries in the hash table are loosely sorted in the following sense that can be exploited to sort the output in linear time. Let pos(m) be the final position of a mer m in the hash table. Then pos(m) = pos(m, i) for some i ∈ [0, max_reprobe − 1]. If pos(m1, 0) + reprobe(max_reprobe) < pos(m2, 0), then pos(m1) < pos(m2). Hence, in order to sort the output, we only need to resolve the proper ordering of the entries within a window of length reprobe(max_reprobe), which is a constant with respect to the size of the hash table, the input size and k. To do so, we create a min-heap of size reprobe(max_reprobe) using the ordering pos(m1, 0) < pos(m2, 0) and lexicographic order to break ties. The elements to write out to disk are read from the head of the heap, and as elements are removed from the heap, new elements are read from the hash table and inserted in the heap.
To parallelize the process of writing to disk, we designed a distributed set of reader–writer locks that optimizes for the common case of hash-table updates and only incurs a significant speed penalty in the rarer case of writing the output. See Appendix Section A.2 in Supplementary Material for details.
3.6 Analysis of running time
It takes a number of operations proportional to αk = 2k ⌈2k/w⌉, where w is the length of a machine word, to compute the matrix-vector multiplication, and the time to insert one k-mer in the hash table is proportional to αk + max_reprobe. To tally nk-mers in the hash takes O(n(αk + max_reprobe)) time. With the choice of quadratic reprobing, the size of the min-heap used to sort the hash table while writing to disk is O(max_reprobe2). Writing n elements to disk involves ninsert and deleteMin operations on the min-heap, hence a cost of O(nlog(max_reprobe)). Hence, creating the hash tables takes time linear in n.
In the case where t intermediary hash tables of size si, 1 ≤ i ≤ t with ∑i=1tsi = n were written to disk, the time to create all t hash tables is O(n(αk + max_reprobe + log(max_reprobe))). The time to merge the t hash tables is O(n log t). If a large amount of memory is available and the number of hash tables created is constant (t = O(1)), then the total runtime is linear in n. In this case, our algorithm is similar to counting sort (Cormen et al., 1990; Seward, 1954) where the array counting the number of occurrences of each element to sort is replaced by a hash table.
At the other extreme, if a small amount of memory is available and the number of hash tables created is proportional to n, then the total runtime is O(n log n). In this later case, the theoretical worst-case performance of the algorithm has degenerated to that of a heap sort (since the time to merge now dominates), although in practice the memory usage and running time will be significantly faster.
4 RESULTS
4.1 Speed and memory usage on Turkey sequencing reads
The memory usage and timing for counting k-mers on sequencing reads of the 1 GB Turkey genome for various levels of coverage are shown in Figures 1 and 2.
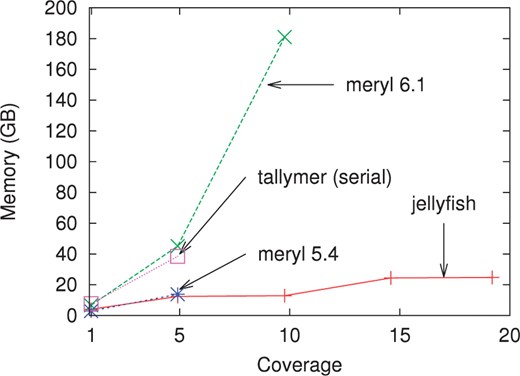
Memory usage for various levels of sequencing coverage on reads generated during the Turkey genome project when counting 22-mers. Except for Tallymer (which is inherently single threaded), all programs were run using 32 threads. The memory usage for the serial and 32-thread versions of Jellyfish is almost identical (results are shown using 32 threads).
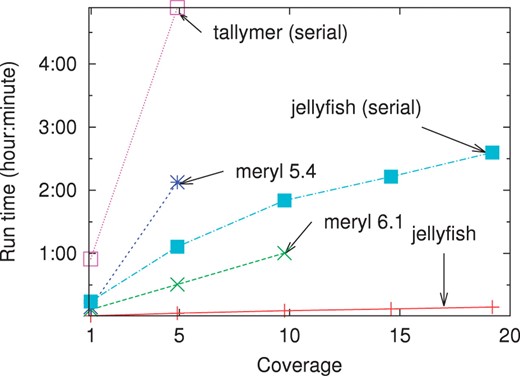
Computation time versus sequencing coverage on reads generated during the Turkey genome project. Except as noted all programs were run using 32 threads.
Jellyfish requires far less memory than the current versions of either Meryl or Tallymer (Fig. 1). The memory usage of Jellyfish is approximately the same for coverage 5× and 10× (or for 15× and 20×) because the size of the hash table is constrained to be a power of two and the same table size of 232 (or 233) entries is used. Tallymer does not support multithreaded operation. When run in serial mode, the memory usage for Jellyfish is almost identical with the usage in multithreaded mode. Meryl version 5.4 contained a software error that prevented it from correctly parsing large input files and was run only up to 5× coverage. At coverage 5×, Jellyfish used only slightly less memory than Meryl 5.4. Meryl version 6.1 ran out of memory for coverage 15× and 20×, and it appears that tradeoffs between speed and memory usage were changed between versions 5.4 and 6.1.
As a comparison, a naïve implementation of a k-mer counter in Python would take > 2 h to count coverage 1×. Jellyfish is also much faster than Meryl and Tallymer (Fig. 2). At coverage 5×, representing approximately 5 GB of sequence, Jellyfish counts 22-mers in under 4 min, while the other approaches take between 30 min and 4.9 h. Jellyfish is also able to count 22-mers at coverages > 10× where the other programs fail or take over 5 h.
The figure in Appendix A.3 shows the impact of varying the mer length k on computation time, showing the contribution of both IO and the actual counting. As k increases from 5 to 30, the counting time stays approximately the same, while the IO time grows significantly because of the larger number of distinct k-mers that must be output. For small values of k (here < 15), Jellyfish uses direct indexing (where there is one entry in the table for each of the 4k possible k-mers). For very low k (here k = 5), the effect of multiple threads trying to increment the same location is compounded and explains the longer counting time than for k = 10.
4.2 Jellyfish's architecture allows for a high degree of parallelism
The CPU and IO usage of Jellyfish on 32 threads while counting 22-mers on coverage 5× of the turkey reads is shown in Figure 3. There are three distinct phases in the trace. First, initialization, when memory is zeroed out and the input file is aggressively preloaded in cache by the operating system. This lasts 14 s. The second phase is active counting which uses 100% of the 32 CPUs available on the machine. Thanks to the lock-free design, the threads almost never wait on each other. The CAS operation is a CPU operation, not a system call that would require an expensive context switch. Therefore, the counting phase is fast and the operating system uses no computational resources during this phase. The final phase is writing, where the results are sorted and written to disk. In this phase, the operations are bounded by IO bandwidth. The default output format, used when creating this trace, is designed to be easy to parse rather than compact. A more compact file format would lead to faster execution time in the third phase.
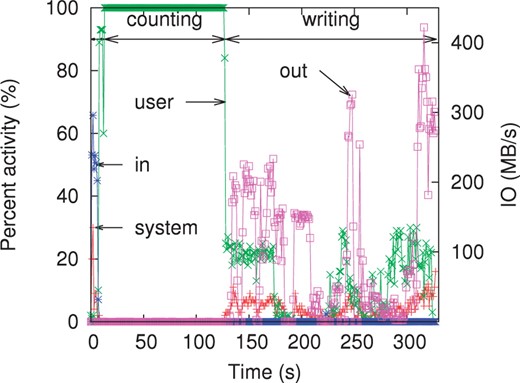
A trace of Jellyfish's CPU usage and IO throughput on counting 22-mers on coverage 5× of the Turkey reads with 32 threads. CPU usage is split into ‘system’ (corresponding to all system calls for memory allocation, read/write from disk, etc.) and ‘user’ (the program). The ‘percent activity’ is a global activity measure over all 32 cores. The IO throughput is split into ‘in’ for input and ‘out’ for output.
Figure 4 shows the speedup obtained when increasing the number of threads used. Again, the k-mers are counted on coverage 5× of the Turkey reads. On the upper curve (labeled ‘no IO’) takes into account only the initialization and counting phases, i.e. only the hash table operations. The lower curve (labeled ‘with IO’) also includes the writing phase where the result is sorted and written to disk. The hash operations have an almost linear speed-up up to 32 threads (the number of cores on the test machine). When including the writing to disk, the speed-up is linear up to four threads and nearly linear up through eight threads. Then, the sorting is done fast enough that the IO bandwidth is the bottleneck (as seen in Fig. 3) and the speed-up levels off.
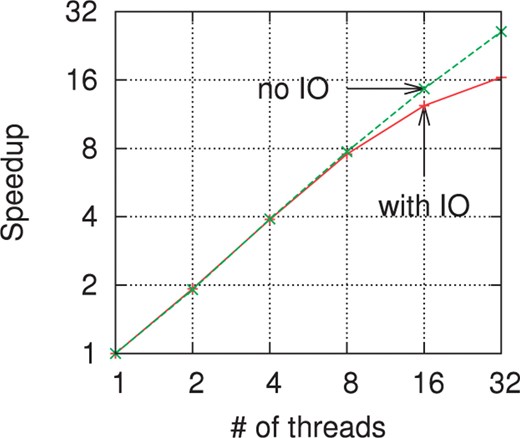
Speedup versus number of threads on coverage 5× of the Turkey reads. On this log–log scale plot, a perfectly linear speedup would correspond to a diagonal line. The ‘no IO’ curves includes only the initialization and counting phase times. The curve marked ‘with IO’ counts the total runtime.
4.3 Timing results for other genomes
Jellyfish is able to process genomes or the reads from recent sequencing projects in only a few minutes. Table 1 contains the timing and memory usage of Jellyfish on several datasets, computed with 32 threads. The number of different k-mers in each dataset is reported as ‘distinct’, and the total number of k-mers is also shown. The timing information for Turkey (M.gallopavo) at coverage 20× is included for comparison. Even on the reads of a very repetitive genome, such as Z.mays, Jellyfish takes <20 min, while computing the suffix array with Tallymer would take ≈24 h.
Performance of Jellyfish on the chromosomes of the human genome and the reads from several sequencing projects
| Organism | Time m:s | RAM GB | Number of 22mers (×106) | ||
|---|---|---|---|---|---|
| Distinct | Total | ||||
| Homo sapiens | 3:33 | 11.8 | 2 351 | 2 861 | |
| Zea mays | 18:14 | 55.9 | 7 161 | 26 653 | |
| Meleagris gallopavo | 9:01 | 24.7 | 5 503 | 19 446 | |
| Drosophila ananassae | 2:19 | 7.35 | 1 197 | 2 936 | |
| Coxiella burnetii | 0:02 | 1.25 | 10.2 | 34.2 | |
| Organism | Time m:s | RAM GB | Number of 22mers (×106) | ||
|---|---|---|---|---|---|
| Distinct | Total | ||||
| Homo sapiens | 3:33 | 11.8 | 2 351 | 2 861 | |
| Zea mays | 18:14 | 55.9 | 7 161 | 26 653 | |
| Meleagris gallopavo | 9:01 | 24.7 | 5 503 | 19 446 | |
| Drosophila ananassae | 2:19 | 7.35 | 1 197 | 2 936 | |
| Coxiella burnetii | 0:02 | 1.25 | 10.2 | 34.2 | |
Performance of Jellyfish on the chromosomes of the human genome and the reads from several sequencing projects
| Organism | Time m:s | RAM GB | Number of 22mers (×106) | ||
|---|---|---|---|---|---|
| Distinct | Total | ||||
| Homo sapiens | 3:33 | 11.8 | 2 351 | 2 861 | |
| Zea mays | 18:14 | 55.9 | 7 161 | 26 653 | |
| Meleagris gallopavo | 9:01 | 24.7 | 5 503 | 19 446 | |
| Drosophila ananassae | 2:19 | 7.35 | 1 197 | 2 936 | |
| Coxiella burnetii | 0:02 | 1.25 | 10.2 | 34.2 | |
| Organism | Time m:s | RAM GB | Number of 22mers (×106) | ||
|---|---|---|---|---|---|
| Distinct | Total | ||||
| Homo sapiens | 3:33 | 11.8 | 2 351 | 2 861 | |
| Zea mays | 18:14 | 55.9 | 7 161 | 26 653 | |
| Meleagris gallopavo | 9:01 | 24.7 | 5 503 | 19 446 | |
| Drosophila ananassae | 2:19 | 7.35 | 1 197 | 2 936 | |
| Coxiella burnetii | 0:02 | 1.25 | 10.2 | 34.2 | |
5 CONCLUSION
Increasingly, practical computation on large collections of genomic sequences requires software which can use parallel computer architectures that are commonly available today. The lock-free operations used in Jellyfish permit the design of truly concurrent data structures that are fast in serial mode and scale almost linearly with the number of processors used.
Jellyfish can tackle k-mer counting on the large datasets available today. As short-read sequencing projects become more common and achieve larger and larger coverage, efficient k-mer counting will become increasingly important. The hash table at the heart of Jellyfish is a versatile and widely used data structure. Proper optimizations make Jellyfish's hash table competitive in both time and space even when compared with other data structures specifically designed for string processing, such as suffix arrays, as implemented in competing k-mer counting packages.
ACKNOWLEDGEMENTS
The authors thank Arthur Delcher for his helpful discussions. We thank Saket Navlakha and Geet Duggal for comments on the manuscript.
Funding: National Science Foundation (grants EF-0849899 and IIS-0812111); National Institutes of Health (grant 1R21AI085376) to C.K. National Science Foundation (grant DMS-0616585); National Institutes of Health (grant 1R01HG0294501).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alex Bateman


{kind=link}
{kind=link}
{kind=link}
{kind=link}