





Abstract
Alignment-free sequence comparison methods can compute the pairwise similarity between a huge number of sequences much faster than sequence-alignment based methods.
We propose a new non-parametric alignment-free sequence comparison method, called K2, based on the Kendall statistics. Comparing to the other state-of-the-art alignment-free comparison methods, K2 demonstrates competitive performance in generating the phylogenetic tree, in evaluating functionally related regulatory sequences, and in computing the edit distance (similarity/dissimilarity) between sequences. Furthermore, the K2 approach is much faster than the other methods. An improved method, , is also proposed, which is able to determine the appropriate algorithmic parameter (length) automatically, without first considering different values. Comparative analysis with the state-of-the-art alignment-free sequence similarity methods demonstrates the superiority of the proposed approaches, especially with increasing sequence length, or increasing dataset sizes.
The K2 and approaches are implemented in the R language as a package and is freely available for open access (http://community.wvu.edu/daadjeroh/projects/K2/K2_1.0.tar.gz).
Supplementary data are available at Bioinformatics online.
1 Introduction
Evaluating the similarity between two sequences is a classical problem that has long been studied in computer science, primarily from the view point of string pattern matching (Adjeroh et al., 2008; Gusfield, 1997). Such similarity measurement has applications in various areas in computational biology, e.g. sequence alignment (Smith and Waterman, 1981), in comparative genomics (Aach et al., 2001), genomic evolution and phylogenetic tree construction and analysis (Cao et al., 1998; Reyes et al., 2000), analysis of regulatory functions (Kantorovitz et al., 2007), rapid search in huge biological sequences (Wandelt and Leser, 2013). Other recent applications include compression and efficient storage of the rapidly expanding genomic datasets (Beal et al., 2016a, b; Deorowicz and Grabowski, 2013; Giancarlo et al., 2012), and resequencing a set of strings given a target string (Kuo et al., 2015), an important step in efficient genome assembly.
Alignment-free sequence comparison methods can compute the similarity between a large number of sequences much faster than alignment-based methods (Vinga and Almeida, 2003; Vinga, 2014). Word analysis of k-length substrings (also called k-mers, k-grams, or k-tuple) from sequences is one approach to improved sequence comparision (Bonham-Carter et al., 2014). Words can be extracted in different ways, and with varying lengths. The most common is to use sliding windows from length 2 to n – 1, where n is the length of sequence (Bauer et al., 2008; Dai et al., 2011; Liu et al., 2006; Qi et al., 2004). Some methods divide a sequence into several even parts (Zhao et al., 2011), while some others have used fixed length substrings, e.g. k = 2 (2-mer) (Shi and Huang, 2012). After extracting the words, different statistical methods can be applied to analyze two sequences for similarity (Li and Wang, 2005; Wang and Zheng, 2008). DMk (Wei et al., 2012) and Category-Position-Frequency (CPF) (Bao et al., 2014) incorporate positions and frequencies of k-mers into feature vectors. DV (Zhao et al., 2011) utilizes distribution vectors from k-mers. Shi (Shi and Huang, 2012) maps a DNA primary sequence into three symbolic sequences and groups these sequences into a twelve-component vector. Wavelet Feature Vector (WFV) converts a sequence into a L-length feature vector by wavelet transform (Bao and Yuan, 2015).
Our approach is more closely related to the D2 statistic, another popular approach for measuring the similarity (or dissimilarity) between two sequences (Bonham-Carter et al., 2014; Song et al., 2014). It was first proposed by Blaisdell (1986). Since then, many variants and improvements have been proposed, such as (Kantorovitz et al., 2007), (Reinert et al., 2009) and (Wan et al., 2010). (Kantorovitz et al., 2007) normalizes the D2 statistic using its mean and standard deviation to improve its detection power (Song et al., 2014). and are two other normalization improvement methods which were proposed in Reinert et al. (2009) and Wan et al. (2010). [also denoted in the literature (Reinert et al., 2009; Song et al., 2014)] uses an approach based on Shepp (1964). According to a recent review (Song et al., 2014), and its variant are generally the best D2 statistical methods for alignment-free comparison of genomic sequences, especially with increasing sequence length. More detailed discussion of the D2-statistic family of algorithms can be founded in Section 6 of the Supplementary Material.
In general, the D2-statistic family of algorithms have a general problem of requiring a quadratic or cubic time complexity, with respect to n or m, the length of the sequences, and k, the size of the substrings being considered. Also, the D2 family of statistics generally makes some assumptions on the distribution of the sequences, for instance, most assumed either a uniform distribution, or a normal distribution, for the symbols in the sequences. This parametric nature of the statistics obviously limits their practical applicability, since practical data, especially for biological sequences (e.g. complete genomes for individuals of the same species, or for related organisms) rarely follow these theoretical distributions. A non-parametric approach to the measurement of sequence similarity is required, one that does not make any assumption on the distribution of the sequences under consideration, and one that is efficient enough to handle the rapidly increasing complexity and data sizes of available biological sequence data.
In this work, we propose a nonparametric approach, K2, which uses the Kendall correlation statistic to estimate the similarity between sequences. The Kendall correlation is a non-parametric method to calculate the correlation between two sets of random variables. We adopt this to measure the similarity among sequences. When compared to the other state-of-the-art alignment-free sequence similarity methods, (e.g. D2, , DMk, DV, CPF, Shi and WFV), K2 demonstrates an improved power in detecting relatedness between sequences, as measured by its ability to generate the correct phylogenetic tree, and to identify functionally related regulatory sequences. The K2 also showed significant correlation with the edit distance, the standard, though time consuming, measure of (similarity/dissimilarity) between sequences. Further, the K2 approach is faster than most of the other methods when k is large, (typically, with ). This places the proposed K2 statistic among the best non-alignment based similarity measures, especially with increasing sequence lengths (n, m), or increasing size of the k-mer. Based on K2, we further propose an improved method, named , which is able to determine a suitable value for k, the k-gram parameter automatically with competitive performance. We have implemented K2 and in the R statistical and graphics environment, and the codes are freely available for open access.
2 Materials and methods
2.1 Kendall statistic
2.2 Optimized computation of Kendall statistics
The time cost to compute , the approximation to the Kendall correlation statistic is , including time to compare each pair between () and (), , where is the number of pairs in and . Christensen (2005) showed an algorithm to calculate in time complexity. It was implemented in Pascal. Lin et al. (2017) recently introduced an algorithm for the related problem of weighted Kendall correlation. In this work, we propose data structures and a new algorithm to compute . Our algorithm also runs in time, but uses a different approach to compute the Kendall statistics. We then apply the algorithm to analyze similarity between a given pair of sequences. More detailed discussion on the improved algorithm for the Kendall Statistics can be found in Section 3.1 of the Supplementary Material.
2.3 The approach
Here, we propose the statistic as a new method for rapid and efficient measurement of biological sequence similarity, without requiring an initial sequence alignment step. The statistic makes use of the above optimized method for computing the Kendall’s correlation between two sequences. Here, the correlation is computed based on the -mer count statistics and between the two sequences. The counts are obtained in time using the suffix array data structure (Adjeroh et al., 2008; Gusfield, 1997; Manber and Myers, 1993), where is the length of input sequence . We describe the steps of the algorithm in the following.
Given two sequences T and P, combine them into one sequence, , after appending an ‘’ at the end of each sequence. The concatenated sequence S is of length .
Build the suffix array (SA) from the combined sequence . And for a given parameter , read all -grams from SA.
Compute the frequency for each -gram using the SA. Here, we use , and to denote the frequency of the -gram in sequences and , respectively. Notice that, both and will be found at essentially the same time, using the SA of the concatenated sequence, .
Order all the () frequencies of -gram pairs by grouping them according to , and then . We get pairs {}, where is the number of distinct -grams from the concatenated sequence , and (Xi, Yi) is the frequency pair of ith ranked k-gram from sequences T and P. Thus, (1) and i < n and (2) when and i < n.
Compute nc, the number of concordant pairs, and nd the number of discordant pairs, for the ranked frequency pairs from sequences T and P. The number of concordant pairs nc is the sum of the number pairs in one of these two conditions: (1) xi < xj and yi < yj; (2) xi > xj and yi > yj, where . Similarly, the number of disconcordant pairs nd, is the sum of the number of pairs in one of the following two conditions: (1) xi < xj and yi > yj; (2) xi > xj and yi < yj, where .
- Calculate the Kendall correlation using the formula:
Return which is the K2 similarity between sequences T and P.
2.4 : improved K2 with automated k value
Similar to the alignment-free methods from the D2 family, the proposed K2 approach depends critically on the length parameter, k. Here, we propose a method to determine the k parameter automatically, without needing to test with all possible values.
2.5 Comparative complexity analysis
The proposed algorithm runs in time, which is a significant improvement in complexity, when compared with the required for computing and other related statistics, or even with the observed improvement that reduces the time to . requires just a one-time run of , using the automatically computed -parameter. This will be practically faster than using , however, the time complexity of still remains the same as in . More detailed discussion can be found in Section 3.2 of the Supplementary Material.
2.6 Experimental design
To test the proposed methods, we performed some experiments using three different datasets. We also compared our experimental results with those from state-of-the-art alignment-free sequence similarity measurement algorithms.
2.6.1 Datasets and environment
We use three sets of biological sequence data for the experiments in this study. The first dataset used is the complete mtDNA sequences from Cao et al. (1998) and Reyes et al. (2000) containing data on 12 proteins encoded in the H strand of mtDNA in 20 eutherian species. The sequence lengths ranged from 16 300 to 17 080 symbols. This dataset is often used to evaluate the similarity of different species, especially using phylogenetic trees. We call this the ‘mtDNA20’ dataset.
The second dataset is 23 whole mitochondrial DNA genomes from different Eukaryotic fish species of the suborder Labroidei, taken from Fischer et al. (2013). We could not locate the sequences for two of the species, namely, P.trewavasae and T.moorii. Thus, though the original work in Fischer et al. (2013) used 25 species, our dataset contained only 23 of the 25 species. The sequence lengths ranged from 16 440 to 17 040 symbols. We call this dataset the ‘Fish23’ dataset.
The third dataset used is the set containing cis-regulatory modules (CRMs) used by Kantorovitz et al. (2007) in their work on identification of functional relationships between cis-regulatory sequences. There are seven sets including 185 CRM sequences, taken from Drosophila melanogaster and Homo sapiens. We call this the ‘CRM185’ dataset. This dataset is available for download at http://veda.cs.uiuc.edu/d2z/publicdata.tar.gz.
The experiments were performed in a PC environment, running Intel i5, 4 cores, with 16 GB RAM and 1 TB HD. and were written using the R Language. For comparison purposes, we also tested several other state-of-the-art alignment-free methods using the same datasets. The algorithm for was from Song et al. (2014), was from Wan et al. (2010), and was from Reinert et al. (2009). They all were implemented using the C language. The method was developed in Perl in the original work of Kantorovitz et al. (2007). We implemented the methods for (Wei et al., 2012), (Bao et al., 2014), (Zhao et al., 2011) and (Shi and Huang, 2012) in R, according to descriptions provided in the respective papers. The codes for , developed in Python in their original work (Bao and Yuan, 2015), were kindly provided by the authors. In our experiments, the parameter corresponds to the length in their work.
2.6.2 Experiment 1
The first experiment aimed at analyzing the general performance of each alignment-free method studied. The experiment compared eleven alignment-free methods, namely, , and and our two proposed methods, and . The experiment was performed on mtDNA20 and Fish23 two datasets.
To evaluate the performance of the algorithms, we consider three performance measures: (i) the Robinson-Foulds (RF) distance (Robinson and Foulds, 1981) which measures the topological distance between the golden reference phylogenetic tree and the phylogenetic tree constructed using a given alignment-free method; (ii) the correlation of the similarity/distance values as determined by the alignment-free method with the standard edit distance; (iii) the computation time required. These performance measures need to be considered both individually and jointly in evaluating algorithms for sequence similarity measurement.
2.6.3 Experiment 2
The second experiment investigated how well the results from the proposed alignment-free methods can capture the similarity between sequences with similar functional roles. For this experiment, we used the related regulatory sequences in the CRM185 dataset, our third dataset. The ‘positive’ set is the set of CRMs that are in the same tissue and/or same developmental stage. The ‘negative’ set is the set chosen from non-coding sequences, which are expected to be unrelated with respect to function. This experiment is designed to predict whether or not any two given sequences are in the ‘positive’ set, using alignment-free methods. First, we compute the similarity between pairwise sequences using alignment-free methods. Next, we rank these pairs based on their similarity, and determine the number of positive pairs and return the accuracy ratio.
3 Results and discussion
3.1 Phylogenetic tree analysis
One way to evaluate the performance of the alignment-free methods is to compare the phylogenetic trees generated using the distance matrix against the known correct (reference) phylogenetic tree for the species in the dataset. In this case, methods that generate trees that have more similarity in structure with the reference tree will be taken to be of better performance.
To compare the similarity/dissimilarity between two trees, we use the Robinson-Foulds(RF) distance (Robinson and Foulds, 1981). The Robinson-Foulds distance (also called the symmetric difference metric) is a well-known approach for measuring the similarity between two trees. [See for example Bansal et al., (2010) and Lu et al., (2017)]. The Robinson-Foulds distance measures the topological distance between two labeled trees essentially by counting the minimum number of elementary operations needed to transform one tree to the other.
For the experiments on the mtDNA20 dataset, and we used the tree published by Cao et al. (1998) as the reference. See also Otu and Sayood (2003). For phylogenetic analysis using the Fish23 dataset, we used the tree published by Fischer et al. (2013) as the reference tree.
3.1.1 mtDNA20 dataset
Table 1 shows the Robinson-Foulds distance between each tree and the reference tree. Each column contains distances of a given alignment-free method with parameter varied from 2–9. The results of three methods without parameter are shown in the last row. The minimum distance in this table is . This minimum was obtained with the method, and it is also present in the column for with parameter =8, 9, and for with parameter =7, 8. The remaining 8 methods are unable to achieve the minimum (best) distance. However, and are able to take the second place with minimum RF distance of 14. and can obtain the minimum RF distance of 16. The distances reported by the other methods, and were far from the minimum distance, hence, were ranked lower. On this dataset, the methods and performed generally better than the others. However, the fact that does not need to try all the possible values from –9, gives it an advantage over the others.
The Robinson-Foulds distance between the reference phylogenetic tree and phylogenetic trees generated using different alignment-free statistical methods (with )
| 2 | 22 | 26 | 26 | 36 | 26 | 18 | 24 | 26 |
| 3 | 24 | 26 | 28 | 34 | 22 | 20 | 22 | 24 |
| 4 | 22 | 20 | 22 | 26 | 22 | 16 | 18 | 24 |
| 5 | 22 | 20 | 16 | 26 | 20 | 16 | 16 | 22 |
| 6 | 24 | 16 | 16 | 24 | 18 | 18 | 16 | 24 |
| 7 | 18 | 14 | 12 | 20 | 14 | 16 | 14 | 24 |
| 8 | 18 | 16 | 12 | 20 | 12 | 16 | 14 | 24 |
| 9 | 16 | 14 | 14 | — | 12 | 18 | 16 | 24 |
| 12 | 20 | 22 | ||||||
| 2 | 22 | 26 | 26 | 36 | 26 | 18 | 24 | 26 |
| 3 | 24 | 26 | 28 | 34 | 22 | 20 | 22 | 24 |
| 4 | 22 | 20 | 22 | 26 | 22 | 16 | 18 | 24 |
| 5 | 22 | 20 | 16 | 26 | 20 | 16 | 16 | 22 |
| 6 | 24 | 16 | 16 | 24 | 18 | 18 | 16 | 24 |
| 7 | 18 | 14 | 12 | 20 | 14 | 16 | 14 | 24 |
| 8 | 18 | 16 | 12 | 20 | 12 | 16 | 14 | 24 |
| 9 | 16 | 14 | 14 | — | 12 | 18 | 16 | 24 |
| 12 | 20 | 22 | ||||||
Note: Results are based on the mtDNA20 dataset (Cao et al., 1998). having automatically determined values, and without varied parameter, they are all reported in the last row for brevity. generated an error at . The bold value 12 here indicates the minimal RF distance. The smaller the RF distance is, the better a method performs.
The Robinson-Foulds distance between the reference phylogenetic tree and phylogenetic trees generated using different alignment-free statistical methods (with )
| 2 | 22 | 26 | 26 | 36 | 26 | 18 | 24 | 26 |
| 3 | 24 | 26 | 28 | 34 | 22 | 20 | 22 | 24 |
| 4 | 22 | 20 | 22 | 26 | 22 | 16 | 18 | 24 |
| 5 | 22 | 20 | 16 | 26 | 20 | 16 | 16 | 22 |
| 6 | 24 | 16 | 16 | 24 | 18 | 18 | 16 | 24 |
| 7 | 18 | 14 | 12 | 20 | 14 | 16 | 14 | 24 |
| 8 | 18 | 16 | 12 | 20 | 12 | 16 | 14 | 24 |
| 9 | 16 | 14 | 14 | — | 12 | 18 | 16 | 24 |
| 12 | 20 | 22 | ||||||
| 2 | 22 | 26 | 26 | 36 | 26 | 18 | 24 | 26 |
| 3 | 24 | 26 | 28 | 34 | 22 | 20 | 22 | 24 |
| 4 | 22 | 20 | 22 | 26 | 22 | 16 | 18 | 24 |
| 5 | 22 | 20 | 16 | 26 | 20 | 16 | 16 | 22 |
| 6 | 24 | 16 | 16 | 24 | 18 | 18 | 16 | 24 |
| 7 | 18 | 14 | 12 | 20 | 14 | 16 | 14 | 24 |
| 8 | 18 | 16 | 12 | 20 | 12 | 16 | 14 | 24 |
| 9 | 16 | 14 | 14 | — | 12 | 18 | 16 | 24 |
| 12 | 20 | 22 | ||||||
Note: Results are based on the mtDNA20 dataset (Cao et al., 1998). having automatically determined values, and without varied parameter, they are all reported in the last row for brevity. generated an error at . The bold value 12 here indicates the minimal RF distance. The smaller the RF distance is, the better a method performs.
Figure 1 shows the reference phylogenetic tree from Cao et al. (1998), and the corresponding tree generated by the proposed approach. Detailed figures for the other methods are presented in the Supplementary Material. To compare different methods, we show the phylogenetic trees constructed using each of the methods. Methods , and depend on the input parameter . For each of these methods, the Supplementary Figure S1 shows the corresponding phylogenetic tree that resulted in the minimum Robinson-Foulds distance with the reference tree. For the method, the value is automatically computed, so, only one tree is generated. The phylogenetic trees from and are not shown in the Supplementary Figure S1 because these trees are far away from the reference tree. See also the RF distances shown in Table 1.
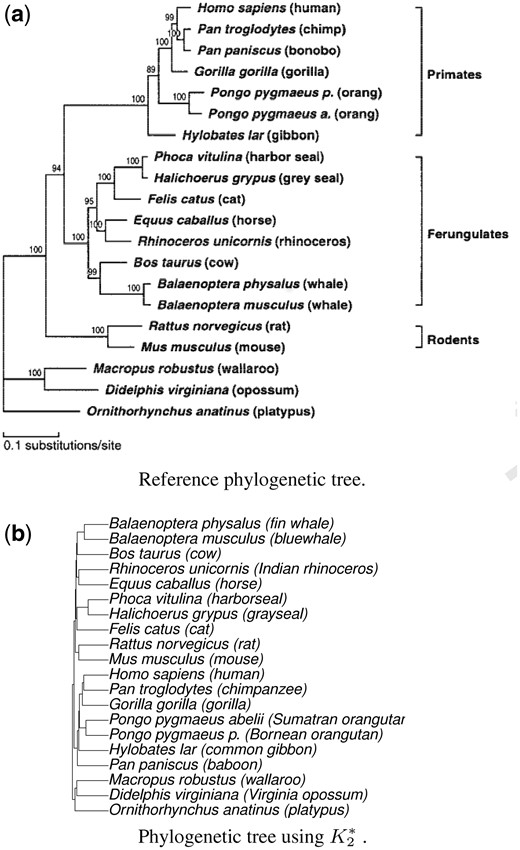
Reference phylogenetic tree from Cao et al. (1998), and the corresponding tree generated using the proposed alignment-free sequence comparison method, using the mtDNA20 dataset
Looking at these figures, we can see that the trees are generally similar to the reference tree, though with some variations. We can observe that and placed horse and white rhinoceros close to each other as expected, however, their parent nodes were wrongly placed, making them much further from say cow than in the reference tree. Also, wrongly placed wallaroo very close to mouse and rat, while had cow much closer to rat and mouse than the reference tree. provided a better result than and , but it also incorrectly placed platypus much closer to rat and mouse. Methods and seem to avoid these problems. One quick way to access the performance of the methods is to compare the minimum number of hops needed to go from one given leaf node (representing a species) to another leaf node on a given tree. The Supplementary Table S3 shows the number of hops for two pairs of species. The Supplementary Figure S1 and Table S3 suggest that the proposed methods and work better than the other methods on the mtDNA20 dataset.
3.1.2 Fish23 dataset
Table 2 shows the Robinson-Foulds distances for the Fish23 dataset. Each column shows distances of one alignment-free method with parameter varied from 2 to 9. The last row shows the results for three methods that did not use varying parameters. The minimum distance in this table is (shown in boldface in the table). Methods and are able to achieve the minimum distance. As with the previous experiment on ‘mtDNA20’, and are worse than the others. Among these methods, only is able to automatically determine an appropriate value. From these results, we conclude that with respect to phylogenetic trees, the is the best amongst all the tested alignment-free methods.
The Robinson-Foulds distance between the reference phylogenetic tree and phylogenetic trees generated using different alignment-free statistical methods (with )
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 32 | 34 | 36 | 40 | 36 | 30 | 32 | 36 |
| 3 | 30 | 30 | 28 | 40 | 26 | 28 | 30 | 30 |
| 4 | 26 | 26 | 30 | 36 | 24 | 22 | 24 | 26 |
| 5 | 24 | 20 | 22 | 38 | 20 | 20 | 20 | 26 |
| 6 | 14 | 10 | 20 | 36 | 12 | 10 | 12 | 32 |
| 7 | 14 | 8 | 14 | 34 | 8 | 12 | 12 | 34 |
| 8 | 8 | 8 | 8 | 34 | 8 | 12 | 14 | 34 |
| 9 | 8 | 10 | 14 | — | 10 | 14 | 16 | 34 |
| 8 | 32 | 34 | ||||||
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 32 | 34 | 36 | 40 | 36 | 30 | 32 | 36 |
| 3 | 30 | 30 | 28 | 40 | 26 | 28 | 30 | 30 |
| 4 | 26 | 26 | 30 | 36 | 24 | 22 | 24 | 26 |
| 5 | 24 | 20 | 22 | 38 | 20 | 20 | 20 | 26 |
| 6 | 14 | 10 | 20 | 36 | 12 | 10 | 12 | 32 |
| 7 | 14 | 8 | 14 | 34 | 8 | 12 | 12 | 34 |
| 8 | 8 | 8 | 8 | 34 | 8 | 12 | 14 | 34 |
| 9 | 8 | 10 | 14 | — | 10 | 14 | 16 | 34 |
| 8 | 32 | 34 | ||||||
Note: Results are based on the Fish23 dataset (Fischer et al., 2013). For brevity, the results for (with automatically determined value), and DV and Shi (both with fixed parameters), are reported in the last row. generated an error at . The bold value 8 here indicates the minimal RF distance. The smaller the RF distance is, the better a method performs.
The Robinson-Foulds distance between the reference phylogenetic tree and phylogenetic trees generated using different alignment-free statistical methods (with )
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 32 | 34 | 36 | 40 | 36 | 30 | 32 | 36 |
| 3 | 30 | 30 | 28 | 40 | 26 | 28 | 30 | 30 |
| 4 | 26 | 26 | 30 | 36 | 24 | 22 | 24 | 26 |
| 5 | 24 | 20 | 22 | 38 | 20 | 20 | 20 | 26 |
| 6 | 14 | 10 | 20 | 36 | 12 | 10 | 12 | 32 |
| 7 | 14 | 8 | 14 | 34 | 8 | 12 | 12 | 34 |
| 8 | 8 | 8 | 8 | 34 | 8 | 12 | 14 | 34 |
| 9 | 8 | 10 | 14 | — | 10 | 14 | 16 | 34 |
| 8 | 32 | 34 | ||||||
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 32 | 34 | 36 | 40 | 36 | 30 | 32 | 36 |
| 3 | 30 | 30 | 28 | 40 | 26 | 28 | 30 | 30 |
| 4 | 26 | 26 | 30 | 36 | 24 | 22 | 24 | 26 |
| 5 | 24 | 20 | 22 | 38 | 20 | 20 | 20 | 26 |
| 6 | 14 | 10 | 20 | 36 | 12 | 10 | 12 | 32 |
| 7 | 14 | 8 | 14 | 34 | 8 | 12 | 12 | 34 |
| 8 | 8 | 8 | 8 | 34 | 8 | 12 | 14 | 34 |
| 9 | 8 | 10 | 14 | — | 10 | 14 | 16 | 34 |
| 8 | 32 | 34 | ||||||
Note: Results are based on the Fish23 dataset (Fischer et al., 2013). For brevity, the results for (with automatically determined value), and DV and Shi (both with fixed parameters), are reported in the last row. generated an error at . The bold value 8 here indicates the minimal RF distance. The smaller the RF distance is, the better a method performs.
The Supplementary Figure S2a shows the phylogenetic tree reported by Fischer et al. (2013) in their original paper using the Fish23 dataset. Similar to the ‘mtDNA20’ experiment, we show the phylogenetic trees generated by the alignment-free methods: , , and . The Supplementary Figure S2(b–h) show the phylogenetic trees with the minimum Robinson-Foulds distance for each method. Supplementary Figure S2a is the reference tree. For our experiments, since we did not have the sequences for P.trewavasae and T.moorii, the pairs N.brichardi, T.duboisi will become neighbors, with parent at node 16 in the original reference tree.
Fish Dataset demonstrates similar trends to the mtDNA20 dataset, see more details in Supplementary Material, Section 4.3.
3.2 Correlation with the edit distance
3.2.1 mtDNA20 dataset
Table 3 shows the Pearson correlation coefficients between the similarity measurements from the different alignment-free methods and the edit distance, using the mtDNA20 dataset. From the table, one can observe that achieved the best result when or achieve the best result () when can reach which is close to the best of . In a word, the method can reach the best accuracy, and is quite competitive. A key advantage of the method is that it is able to select parameter automatically and quickly. However, considering the may need to try all possible values to determine the best (9 in this case), the slight performance disadvantage ( versus ) of becomes even less significant, especially when data volume is huge. See more detailed analysis in Supplementary Material, Section 4.1.1.
Pearson correlation coefficient between the similarity/distance measure from different alignment-free statistical methods and the edit distance
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | −0.45 | −0.51 | −0.55 | 0.02 | −0.56 | 0.67 | 0.62 | 0.57 |
| 3 | −0.48 | −0.60 | −0.74 | 0.10 | −0.73 | 0.68 | 0.66 | 0.62 |
| 4 | −0.53 | −0.71 | −0.86 | −0.74 | −0.82 | 0.70 | 0.71 | 0.63 |
| 5 | −0.61 | −0.79 | −0.91 | −0.81 | −0.89 | 0.78 | 0.77 | 0.72 |
| 6 | −0.77 | −0.87 | −0.92 | −0.83 | −0.92 | 0.84 | 0.86 | 0.68 |
| 7 | −0.87 | −0.91 | −0.92 | −0.84 | −0.92 | 0.87 | 0.89 | 0.68 |
| 8 | −0.90 | −0.92 | −0.91 | −0.84 | −0.93 | 0.85 | 0.89 | 0.66 |
| 9 | −0.91 | −0.91 | −0.91 | — | −0.95 | 0.85 | 0.87 | 0.67 |
| −0.94 | 0.70 | 0.68 |
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | −0.45 | −0.51 | −0.55 | 0.02 | −0.56 | 0.67 | 0.62 | 0.57 |
| 3 | −0.48 | −0.60 | −0.74 | 0.10 | −0.73 | 0.68 | 0.66 | 0.62 |
| 4 | −0.53 | −0.71 | −0.86 | −0.74 | −0.82 | 0.70 | 0.71 | 0.63 |
| 5 | −0.61 | −0.79 | −0.91 | −0.81 | −0.89 | 0.78 | 0.77 | 0.72 |
| 6 | −0.77 | −0.87 | −0.92 | −0.83 | −0.92 | 0.84 | 0.86 | 0.68 |
| 7 | −0.87 | −0.91 | −0.92 | −0.84 | −0.92 | 0.87 | 0.89 | 0.68 |
| 8 | −0.90 | −0.92 | −0.91 | −0.84 | −0.93 | 0.85 | 0.89 | 0.66 |
| 9 | −0.91 | −0.91 | −0.91 | — | −0.95 | 0.85 | 0.87 | 0.67 |
| −0.94 | 0.70 | 0.68 |
Note: Reports are for the mtDNA20 dataset. having automatically determined values, and without varied parameter, they are all reported in the last row for brevity. generated an error at . The bold values indicate the biggest absolute value of Pearson correlation coefficient for different k values. The bigger an absolute value, the better a method performs.
Pearson correlation coefficient between the similarity/distance measure from different alignment-free statistical methods and the edit distance
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | −0.45 | −0.51 | −0.55 | 0.02 | −0.56 | 0.67 | 0.62 | 0.57 |
| 3 | −0.48 | −0.60 | −0.74 | 0.10 | −0.73 | 0.68 | 0.66 | 0.62 |
| 4 | −0.53 | −0.71 | −0.86 | −0.74 | −0.82 | 0.70 | 0.71 | 0.63 |
| 5 | −0.61 | −0.79 | −0.91 | −0.81 | −0.89 | 0.78 | 0.77 | 0.72 |
| 6 | −0.77 | −0.87 | −0.92 | −0.83 | −0.92 | 0.84 | 0.86 | 0.68 |
| 7 | −0.87 | −0.91 | −0.92 | −0.84 | −0.92 | 0.87 | 0.89 | 0.68 |
| 8 | −0.90 | −0.92 | −0.91 | −0.84 | −0.93 | 0.85 | 0.89 | 0.66 |
| 9 | −0.91 | −0.91 | −0.91 | — | −0.95 | 0.85 | 0.87 | 0.67 |
| −0.94 | 0.70 | 0.68 |
| k | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2 | −0.45 | −0.51 | −0.55 | 0.02 | −0.56 | 0.67 | 0.62 | 0.57 |
| 3 | −0.48 | −0.60 | −0.74 | 0.10 | −0.73 | 0.68 | 0.66 | 0.62 |
| 4 | −0.53 | −0.71 | −0.86 | −0.74 | −0.82 | 0.70 | 0.71 | 0.63 |
| 5 | −0.61 | −0.79 | −0.91 | −0.81 | −0.89 | 0.78 | 0.77 | 0.72 |
| 6 | −0.77 | −0.87 | −0.92 | −0.83 | −0.92 | 0.84 | 0.86 | 0.68 |
| 7 | −0.87 | −0.91 | −0.92 | −0.84 | −0.92 | 0.87 | 0.89 | 0.68 |
| 8 | −0.90 | −0.92 | −0.91 | −0.84 | −0.93 | 0.85 | 0.89 | 0.66 |
| 9 | −0.91 | −0.91 | −0.91 | — | −0.95 | 0.85 | 0.87 | 0.67 |
| −0.94 | 0.70 | 0.68 |
Note: Reports are for the mtDNA20 dataset. having automatically determined values, and without varied parameter, they are all reported in the last row for brevity. generated an error at . The bold values indicate the biggest absolute value of Pearson correlation coefficient for different k values. The bigger an absolute value, the better a method performs.
Similar results were observed using the Fish23 dataset. These have been included in Section 4.1.2 of Supplementary Material.
3.3 Practical running time
We compare the running time of eleven methods, [9 earlier approaches ( and ) and the two proposed methods ( and )].
3.3.1 mtDNA20 dataset
Table 4 shows a comparison of the running time for eleven methods using the first dataset (mtDNA20 dataset) from Cao et al. (1998). Figure 2 plots the corresponding running times. The time for is 3.07 s, time for =2.33 s and time for =1.37 s which are not plotted in the figure. When generates a runtime error, thus, we could not obtain a result for this case.
Practical running time (in seconds) using alignment-free methods on the mtDNA20 dataset
| 2 | 0.02 | 0.05 | 0.05 | 1.55 | 0.41 | 3.64 | 13.23 | 0.004 |
| 3 | 0.03 | 0.05 | 0.07 | 1.56 | 0.45 | 4.90 | 14.02 | 0.008 |
| 4 | 0.08 | 0.11 | 0.15 | 1.61 | 0.57 | 5.91 | 15.63 | 0.020 |
| 5 | 0.20 | 0.34 | 0.5 | 1.76 | 1.94 | 7.06 | 16.82 | 0.088 |
| 6 | 0.56 | 1.29 | 2 | 2.35 | 2.22 | 9.78 | 16.58 | 0.884 |
| 7 | 1.26 | 4.91 | 7 | 5.38 | 3.17 | 18.09 | 16.43 | 7.768 |
| 8 | 2.40 | 18.18 | 25 | 19.19 | 3.63 | 40.13 | 16.82 | 38.3 |
| 9 | 4.58 | 70.28 | 99 | — | 4.34 | 92.10 | 17.05 | 347.1 |
| 3.07 | 2.33 | 1.37 |
| 2 | 0.02 | 0.05 | 0.05 | 1.55 | 0.41 | 3.64 | 13.23 | 0.004 |
| 3 | 0.03 | 0.05 | 0.07 | 1.56 | 0.45 | 4.90 | 14.02 | 0.008 |
| 4 | 0.08 | 0.11 | 0.15 | 1.61 | 0.57 | 5.91 | 15.63 | 0.020 |
| 5 | 0.20 | 0.34 | 0.5 | 1.76 | 1.94 | 7.06 | 16.82 | 0.088 |
| 6 | 0.56 | 1.29 | 2 | 2.35 | 2.22 | 9.78 | 16.58 | 0.884 |
| 7 | 1.26 | 4.91 | 7 | 5.38 | 3.17 | 18.09 | 16.43 | 7.768 |
| 8 | 2.40 | 18.18 | 25 | 19.19 | 3.63 | 40.13 | 16.82 | 38.3 |
| 9 | 4.58 | 70.28 | 99 | — | 4.34 | 92.10 | 17.05 | 347.1 |
| 3.07 | 2.33 | 1.37 |
Note: Results for with automatically determined values, and with fixed values, are reported in the last row. generated an error at . The bold values shown the smallest running time (the fastest method) for different k values.
Practical running time (in seconds) using alignment-free methods on the mtDNA20 dataset
| 2 | 0.02 | 0.05 | 0.05 | 1.55 | 0.41 | 3.64 | 13.23 | 0.004 |
| 3 | 0.03 | 0.05 | 0.07 | 1.56 | 0.45 | 4.90 | 14.02 | 0.008 |
| 4 | 0.08 | 0.11 | 0.15 | 1.61 | 0.57 | 5.91 | 15.63 | 0.020 |
| 5 | 0.20 | 0.34 | 0.5 | 1.76 | 1.94 | 7.06 | 16.82 | 0.088 |
| 6 | 0.56 | 1.29 | 2 | 2.35 | 2.22 | 9.78 | 16.58 | 0.884 |
| 7 | 1.26 | 4.91 | 7 | 5.38 | 3.17 | 18.09 | 16.43 | 7.768 |
| 8 | 2.40 | 18.18 | 25 | 19.19 | 3.63 | 40.13 | 16.82 | 38.3 |
| 9 | 4.58 | 70.28 | 99 | — | 4.34 | 92.10 | 17.05 | 347.1 |
| 3.07 | 2.33 | 1.37 |
| 2 | 0.02 | 0.05 | 0.05 | 1.55 | 0.41 | 3.64 | 13.23 | 0.004 |
| 3 | 0.03 | 0.05 | 0.07 | 1.56 | 0.45 | 4.90 | 14.02 | 0.008 |
| 4 | 0.08 | 0.11 | 0.15 | 1.61 | 0.57 | 5.91 | 15.63 | 0.020 |
| 5 | 0.20 | 0.34 | 0.5 | 1.76 | 1.94 | 7.06 | 16.82 | 0.088 |
| 6 | 0.56 | 1.29 | 2 | 2.35 | 2.22 | 9.78 | 16.58 | 0.884 |
| 7 | 1.26 | 4.91 | 7 | 5.38 | 3.17 | 18.09 | 16.43 | 7.768 |
| 8 | 2.40 | 18.18 | 25 | 19.19 | 3.63 | 40.13 | 16.82 | 38.3 |
| 9 | 4.58 | 70.28 | 99 | — | 4.34 | 92.10 | 17.05 | 347.1 |
| 3.07 | 2.33 | 1.37 |
Note: Results for with automatically determined values, and with fixed values, are reported in the last row. generated an error at . The bold values shown the smallest running time (the fastest method) for different k values.
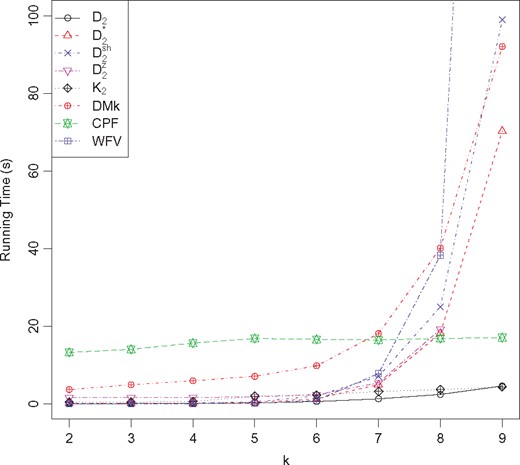
Time cost comparison for and with parameter varying from 2 to 9, using the mtDNA20 dataset. Results for =3.07 s, =2.33 s and =1.37 s are not shown in the figure for clarity
First, consider the methods that use varied values. When , the approach is the fastest among all methods. When the parameter increases, the running time of increases rapidly, much quicker than all the others. When requires approximately 2.45, 10.55 and 109.5-fold time increases, respectively, when compared with . Therefore, in terms of running time, is the better choice than the other methods, with less running time and higher accuracy when . The method with RF distances (26, 24 and 22) shown in Table 1 did not perform well.
Consider and , the two methods that achieved the best results with RF distance = 12 in Table 1. reaches its best performance when reaches its best performance when . When requires approximately 2, 8 and 25 fold time increases, respectively, when compared with . Therefore, in terms of combining with running time and accuracy, is the better choice than .
Now consider and which do not use varying values. requires 3.07 s to execute. and are relatively faster with 2.33 and 1.37 s respectively. However, generated a much lower RF distance—see Table 1. is slower than the other methods (i.e. ) with , and faster than the other methods with . We can also observe from the results discussed earlier that, for this dataset, the best performance for the other methods were recorded at k ≥ 6. See Supplementary Figure S1 and Table 1. Clearly, since does not need to search for the best value (i.e. it is executed for just one value), it is overall faster than the other methods, without degrading the accuracy. This is important, considering the increasingly huge volumes of data involved in most applications of these techniques. In fact, the primary motivation for the alignment-free methods is their rapid processing speed, when compared with alignment-based methods.
Results on running time using the Fish23 dataset is provided in the Supplementary Material.
With respect to running time, we can identify two key points from our experiments: (i) the running time for , and increases rapidly with increasing . The running time for is approximately linear with respect to the sequence length. (ii) Comparing and is more practical, since it can determine the value automatically, and has a competitive performance.
3.4 Evaluation on functionally related regulatory sequences
While the alignment-free methods could be generally fast, an important consideration is whether they can identify similarities between sequences that are functionally related. Of course, this can only be possible if the sequences share some similar patterns. To evaluate this aspect of performance, we consider to what extent the alignment-free similarity measures are able to capture the similarities between sequences from the same anatomic regions of the same species. For this experiment, we used the third dataset—CRM185 dataset, the regulatory sequences from Kantorovitz et al. (2007). We compare our proposed methods and against and , and . Table 5 shows the results. In the table, the result for is taken from the original work of Kantorovitz et al. (2007). For and , the table shows the best results with values in the range . For method, we also tested with .
The performance of popular alignment-free sequence similarity methods in capturing functional relatedness
| Species | Dataset | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fly | Blastoderm | 0.73 | 0.92(4) | 0.85(2) | 0.82(2) | 0.82(6) | 0.79 | 0.83(3) | 0.84(4) | 0.79(5) | 0.72 | 0.7 |
| Fly | PNS | 0.62 | 0.60(5) | 0.63(3) | 0.64(4) | 0.64(3) | 0.56 | 0.62(4) | 0.61(4) | 0.63(3) | 0.58 | 0.55 |
| Fly | Tracheal | 0.75 | 0.75(4) | 0.72(4) | 0.69(4) | 0.69(4) | 0.75 | 0.73(3) | 0.75(4) | 0.70(5) | 0.7 | 0.71 |
| Fly | Eye | 0.58 | 0.69(3) | 0.61(2) | 0.63(3) | 0.60(3) | 0.69 | 0.63(5) | 0.63(4) | 0.64(3) | 0.62 | 0.59 |
| Human | Muscle | 0.83 | 0.88(4) | 0.83(5) | 0.83(5) | 0.86(6) | 0.81 | 0.84(3) | 0.82(4) | 0.81(5) | 0.76 | 0.79 |
| Human | Liver | 0.69 | 0.83(2) | 0.88(2) | 0.78(6) | 0.73(4) | 0.69 | 0.82(2) | 0.84(5) | 0.80(3) | 0.8 | 0.76 |
| Human | HBB | 0.64 | 0.65(3) | 0.58(3) | 0.53(2) | 0.59(4) | 0.66 | 0.57(2) | 0.60(3) | 0.61(4) | 0.56 | 0.52 |
| Species | Dataset | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fly | Blastoderm | 0.73 | 0.92(4) | 0.85(2) | 0.82(2) | 0.82(6) | 0.79 | 0.83(3) | 0.84(4) | 0.79(5) | 0.72 | 0.7 |
| Fly | PNS | 0.62 | 0.60(5) | 0.63(3) | 0.64(4) | 0.64(3) | 0.56 | 0.62(4) | 0.61(4) | 0.63(3) | 0.58 | 0.55 |
| Fly | Tracheal | 0.75 | 0.75(4) | 0.72(4) | 0.69(4) | 0.69(4) | 0.75 | 0.73(3) | 0.75(4) | 0.70(5) | 0.7 | 0.71 |
| Fly | Eye | 0.58 | 0.69(3) | 0.61(2) | 0.63(3) | 0.60(3) | 0.69 | 0.63(5) | 0.63(4) | 0.64(3) | 0.62 | 0.59 |
| Human | Muscle | 0.83 | 0.88(4) | 0.83(5) | 0.83(5) | 0.86(6) | 0.81 | 0.84(3) | 0.82(4) | 0.81(5) | 0.76 | 0.79 |
| Human | Liver | 0.69 | 0.83(2) | 0.88(2) | 0.78(6) | 0.73(4) | 0.69 | 0.82(2) | 0.84(5) | 0.80(3) | 0.8 | 0.76 |
| Human | HBB | 0.64 | 0.65(3) | 0.58(3) | 0.53(2) | 0.59(4) | 0.66 | 0.57(2) | 0.60(3) | 0.61(4) | 0.56 | 0.52 |
Note: Numbers in brackets indicate the value that produced the best results for the given method. Results are based on the CRM185 dataset. The bold values shown the best methods on a data set without considering K2* and with considering K2*.
The performance of popular alignment-free sequence similarity methods in capturing functional relatedness
| Species | Dataset | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fly | Blastoderm | 0.73 | 0.92(4) | 0.85(2) | 0.82(2) | 0.82(6) | 0.79 | 0.83(3) | 0.84(4) | 0.79(5) | 0.72 | 0.7 |
| Fly | PNS | 0.62 | 0.60(5) | 0.63(3) | 0.64(4) | 0.64(3) | 0.56 | 0.62(4) | 0.61(4) | 0.63(3) | 0.58 | 0.55 |
| Fly | Tracheal | 0.75 | 0.75(4) | 0.72(4) | 0.69(4) | 0.69(4) | 0.75 | 0.73(3) | 0.75(4) | 0.70(5) | 0.7 | 0.71 |
| Fly | Eye | 0.58 | 0.69(3) | 0.61(2) | 0.63(3) | 0.60(3) | 0.69 | 0.63(5) | 0.63(4) | 0.64(3) | 0.62 | 0.59 |
| Human | Muscle | 0.83 | 0.88(4) | 0.83(5) | 0.83(5) | 0.86(6) | 0.81 | 0.84(3) | 0.82(4) | 0.81(5) | 0.76 | 0.79 |
| Human | Liver | 0.69 | 0.83(2) | 0.88(2) | 0.78(6) | 0.73(4) | 0.69 | 0.82(2) | 0.84(5) | 0.80(3) | 0.8 | 0.76 |
| Human | HBB | 0.64 | 0.65(3) | 0.58(3) | 0.53(2) | 0.59(4) | 0.66 | 0.57(2) | 0.60(3) | 0.61(4) | 0.56 | 0.52 |
| Species | Dataset | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fly | Blastoderm | 0.73 | 0.92(4) | 0.85(2) | 0.82(2) | 0.82(6) | 0.79 | 0.83(3) | 0.84(4) | 0.79(5) | 0.72 | 0.7 |
| Fly | PNS | 0.62 | 0.60(5) | 0.63(3) | 0.64(4) | 0.64(3) | 0.56 | 0.62(4) | 0.61(4) | 0.63(3) | 0.58 | 0.55 |
| Fly | Tracheal | 0.75 | 0.75(4) | 0.72(4) | 0.69(4) | 0.69(4) | 0.75 | 0.73(3) | 0.75(4) | 0.70(5) | 0.7 | 0.71 |
| Fly | Eye | 0.58 | 0.69(3) | 0.61(2) | 0.63(3) | 0.60(3) | 0.69 | 0.63(5) | 0.63(4) | 0.64(3) | 0.62 | 0.59 |
| Human | Muscle | 0.83 | 0.88(4) | 0.83(5) | 0.83(5) | 0.86(6) | 0.81 | 0.84(3) | 0.82(4) | 0.81(5) | 0.76 | 0.79 |
| Human | Liver | 0.69 | 0.83(2) | 0.88(2) | 0.78(6) | 0.73(4) | 0.69 | 0.82(2) | 0.84(5) | 0.80(3) | 0.8 | 0.76 |
| Human | HBB | 0.64 | 0.65(3) | 0.58(3) | 0.53(2) | 0.59(4) | 0.66 | 0.57(2) | 0.60(3) | 0.61(4) | 0.56 | 0.52 |
Note: Numbers in brackets indicate the value that produced the best results for the given method. Results are based on the CRM185 dataset. The bold values shown the best methods on a data set without considering K2* and with considering K2*.
The bold items are the best results on the dataset comparing different methods, while excluding . From Table 5, reported five best results out of seven using the CRM185 dataset. and reported one best result each. demonstrates competitive performance with the other methods. When we take into consideration, we can observe that it gets three best results out of seven datasets. and get one best result of seven cases, and are best on two cases, and was best on four cases. In general, the proposed and methods provide the overall best performance on this problem.
4 Conclusion
The problem of sequence similarity measurement is critical to several important applications in huge volume genomic sequence analysis. We proposed a novel non-parametric algorithm for alignment-free measurement of relatedness between sequences, using the statistics of -grams in the sequences. is a non-parametric approach based on the Kendall correlation statistic to estimate the dissimilarity(/similarity) of sequences.
Compared with other state-of-the-art alignment-free comparison methods ( and ), demonstrates comparable or better performance, in phylogenetic analysis, in generating (similarity/dissimilarity) measures that correlate with the edit distance among a large number of sequences, and in capturing functional relatedness between sequences. Further, the approach is faster than the other methods when
We also introduced , an improved version of that is able to automatically determine the suitable value, thus eliminating the need to search for all possible values (for the -grams), potentially from to produced the best overall results, with respect to both efficiency and accuracy. Along with competitive performance in measuring the similarity between sequences, its speed makes it practical, an important consideration given the increasingly huge datasets in various applications of alignment-free methods.
Acknowledgement
A shorter version of this paper was presented at IEEE BIBM’2016, Lin et al. (2016).
Funding
This work was supported by the Chinese National Natural Science Foundation (Grant No. 61472082), Natural Science Foundation of Fujian Province of China (Grant No. 2014J01220), the US National Science Foundation (Grant No. IIS-1552860) and Scientific Research Innovation Team Construction Program of Fujian Normal University (Grant No. IRTL1702).
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}