





Abstract
Tumor genome sequencing offers great promise for guiding research and therapy, but spurious variant calls can arise from multiple sources. Mouse contamination can generate many spurious calls when sequencing patient-derived xenografts. Paralogous genome sequences can also generate spurious calls when sequencing any tumor. We developed a BLAST-based algorithm, Mouse And Paralog EXterminator (MAPEX), to identify and filter out spurious calls from both these sources.
When calling variants from xenografts, MAPEX has similar sensitivity and specificity to more complex algorithms. When applied to any tumor, MAPEX also automatically flags calls that potentially arise from paralogous sequences. Our implementation, mapexr, runs quickly and easily on a desktop computer. MAPEX is thus a useful addition to almost any pipeline for calling genetic variants in tumors.
The mapexr package for R is available at https://github.com/bmannakee/mapexr under the MIT license.
Supplementary data are available at Bioinformatics online.
1 Introduction
Molecular characterization of tumors is an important tool in cancer research, and the large-scale sequencing of cancer genomes has led to a deeper understanding of many aspects of the biology of cancer (Stratton, 2011). It is now common to sequence tumors from large cohorts of patients, as well as patient-derived xenograft (PDX) models from individual patients. Such sequencing enables identification of mutational signatures (Alexandrov et al., 2013), functionally important variants (Ding et al., 2012) and evolutionary history of the tumor (Carter et al., 2012; Nik-Zainal et al., 2012). These genetic features are relevant in evaluating etiological mechanisms (Yachida et al., 2010), prognostic subtypes (Park et al., 2010; Shah et al., 2009), and acquired therapeutic resistance (Witkiewicz et al., 2015). All these applications of tumor sequencing depend on sensitive and specific characterization of low-frequency mutations, and as a result may be biased by spurious variant calls. Here, we focus on two specific sources of spurious calls, mouse cell contamination in PDX tumors and mis-alignment of paralogous sequences.
PDX models serve as avatars for individual patient tumors when studying intra-tumor heterogeneity and metastasis and when screening anti-cancer compounds (Allaway et al., 2016; Bruna et al., 2016; Dawson et al., 2012; Day et al., 2015; Knudsen et al., 2017). The primary difficulty in sequencing these models is that mouse stroma is present in all PDX tumors. The high genetic similarity between mouse and human then causes bias when variants are called using bioinformatic pipelines originally developed for primary tumors (Rossello et al., 2013; Tso et al., 2014). Several methods have been developed to facilitate the accurate calling of variants in PDX models. Experimentally, human-specific fluorescence tags can be used to label and isolate human cells prior to DNA extraction (Schneeberger et al., 2016). Bioinformatically, sequence reads can be aligned to both human and mouse reference genomes, either separately (Conway et al., 2012; Khandelwal et al., 2017) or simultaneously (Bruna et al., 2016), to filter out mouse reads prior to variant calling. Although these approaches greatly improve the reliability of variant calls from PDX models, they entail substantial experimental or bioinformatic burdens. Here, we describe a light-weight filtering algorithm that achieves equivalent reliability and can be easily added to standard bioinformatic pipelines, because it uses the same reference genome for alignment as primary tumors.
Many human genes have highly similar paralogous sequences in the genome. Spurious variant calls arising from such paralogs have been recognized as an important source of false positives in the study of rare disease-associated germline variants (Jia et al., 2012; Mandelker et al., 2016; Ng et al., 2010; Zhou et al., 2015). Similarly, paralogs have led to false positives in the study of cancer, including TUBB in non-small cell lung cancer (Kelley et al., 2001), PIK3CA in hepatocellular carcinoma (Müller et al., 2007; Tanaka et al., 2006) and MLL3 in myelodysplastic syndrome (Bowler et al., 2014). To address the paralog problem, some variant callers, such as MuTect2 [currently in beta but included in the Genome Analysis Toolkit (GATK; McKenna et al., 2010)], filter clustered variants, which often result from mis-alignment of paralogous sequences. Many labs also keep lists of suspect genes that tend to suffer from paralog problems and simply ignore any variants called in these genes. These approaches introduce their own biases. Our approach automatically identifies potential spurious calls from paralogs and enables flexible evidence-based filtering.
Here, we fully describe and characterize MAPEX (the Mouse And Paralog EXterminator), a BLASTN-based algorithm for filtering variants that was previously introduced by Knudsen et al. (2017). We also present mapexr, a fast and light-weight implementation in R. The MAPEX algorithm is aimed at three use cases.
Labs that sequence PDX tumors using services that align to the human reference can easily and accurately filter mouse contamination with mapexr.
Bioinformatically sophisticated labs could align against both the human and mouse genomes to use other filtering approaches, but mapexr enables additional variant-level assessment of results.
Any tumor genomics lab can use mapexr as a light-weight approach to identify potentially spurious variants created by paralogous sequences.
2 Approach
2.1 Workflow
The MAPEX algorithm is a post-variant-calling filter designed to fit into a standard tumor variant calling pipeline and flag variants which may arise from mis-alignment of mouse reads or from paralogous sequences (Fig. 1). The input for MAPEX is a BAM file containing tumor reads aligned to the human reference genome and a variant callset generated from that alignment. Variant-supporting reads are then BLASTed against the appropriate reference genome(s). Variants are scored by the fraction of supporting reads that align to the called site of the variant in the human genome.
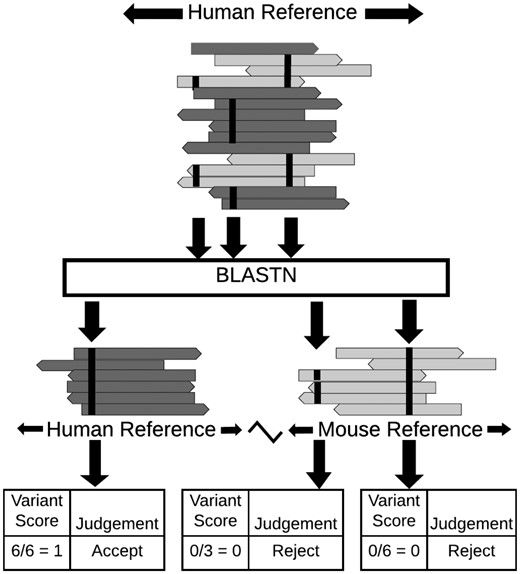
Illustration of MAPEX applied to a PDX sample. MAPEX begins with variants called from tumor reads aligned to the human genome. For each variant, the supporting reads are BLASTed against the combined human and mouse reference genomes. Variants are then scored by the fraction of supporting reads that align to the called site of the variant in the human genome
2.2 Algorithm
Each read supporting a variant is BLASTed against the appropriate reference genome for the application. For PDX applications, this is the combined human/mouse reference, and for primary tumor applications, this is just the human reference. The best hit for each read is determined by bit score. Reads for which the best hit overlaps the called variant location are classified as ‘on target’ and assigned a score of 1. Reads for which the best hit is a different region of the human genome or a region of the mouse genome are classified as ‘off target’ or ‘mouse,’ respectively, and assigned a score of 0. Reads from genes with close paralogs in the human genome may generate multiple best hits (ties). In this case, the read score is averaged over all best hits, and the read is classified based on the most common result from the best hits. Each variant is then assigned a score that is the average score of all reads supporting that variant and is classified based on the most common classification of the supporting reads.
2.3 Implementation
We have implemented the MAPEX algorithm as an R package (mapexr). The package leverages the Bioconductor packages Rsamtools, GenomicAlignments and GenomicRanges for fast and memory-efficient BAM file handling and read sequence extraction (Lawrence et al., 2013; Morgan et al., 2017). The package requires a local BLASTN installation and a BLAST database constructed from either a combined human/mouse reference genome or a human reference genome, depending on the application.
3 Materials and methods
3.1 Samples
To characterize the performance of MAPEX, we used whole exome sequence trimmed fastq reads obtained from pancreatic ductal adenocarcinoma (PDAC) samples described previously by Knudsen et al. (2017) (PDX) and Witkiewicz et al. (2015) (primary). For the PDX analysis, we analyzed a total of 34 PDXs derived from 9 primary tumors, sequenced to mean coverage depth of 124x. For the paralog analysis, we analyzed 93 primary tumors sequenced to a mean coverage depth of 40x.
3.2 Alignments and variant callers
All alignments were done using bwa-mem with default parameter settings (Li and Durbin, 2009). For initial variant calling, we aligned all reads in the samples to the human reference genome GRCh37. We then called variants using MuTect version 1.1.1 (Cibulskis et al., 2013), MuTect2 (as part of the GATK version 3.6, McKenna et al., 2010) and Varscan 2 (Koboldt et al., 2012), all with default parameters. MuTect 1 and 2 variant calls were used without any post-filtering, but for Varscan 2 we used the built-in processSomatic and fpfilter functions with default parameters to generate a set of high-confidence variant calls. Variants were annotated with Oncotator (Ramos et al., 2015) and the annotation database oncotator_v1_ds_April052016. We considered only non-synonymous single nucleotide variants when comparing between methods. For paralog filtering, we used a conservative variant score cutoff of 0.8.
For comparison with Bruna et al. (2016), we aligned reads to a combined human/mouse reference genome GRCh37/mm9 and called variants using MuTect 1.1.1. We calculated the fraction of mouse contamination using the method described in Bruna et al. (2016). Briefly, they generated data comparing the fraction of mouse cells in a sample with the fraction of total reads aligned to the mouse portion of a combined reference genome. We used this data to fit a LOESS regression model for contamination fraction versus fraction aligned, and used this to predict mouse contamination based on the fraction of reads aligned to the mouse genome in our samples.
For comparison with bamcmp (Khandelwal et al., 2017), we aligned reads separately to the human and mouse reference genomes and ran bamcmp with default parameters. The output of bamcmp includes alignment files for reads that aligned to only the human reference and that aligned to both references but with a higher human alignment score. We merged these two alignments, performed indel re-alignment and base score re-calibration using the GATK, and used the merged alignment to call variants with Mutect version 1.1.1. All scripts (doi: 10.5281/zenodo.1112101) and the version of mapexr (doi: 10.5281/zenodo.1112234) used to conduct the analysis have been archived with Zenodo.
4 Results and discussion
4.1 Methodological
MAPEX is a light-weight filtering algorithm that adds little overhead or complexity to existing tumor variant-calling pipelines. The runtime for mapexr is linear in the number of variants to be filtered, processing roughly 250 variants per minute on a 4-core machine (Supplementary Fig. S1).
MAPEX has only one tunable parameter, the minimum mapping quality score required for a variant read. The default minimum score is 1, which includes all reads with an unambiguous best mapping. In pipelines in which a minimum mapping quality score is used for variant calling, that score should also be supplied to mapexr, to prevent evaluating reads that were not used by the variant caller. The output from mapexr is an R data frame with four columns—chromosome, start location, variant score and variant classification—and one row for each variant evaluated. Users may also optionally provide a file path to mapexr which will generate a tab-delimited file with BLAST results and scores at the read level. The user can choose the variant score threshold used to classify variants as human- or mouse-derived. Here, we use a threshold of 0.5, so that a variant is flagged as spurious if less than half of the supporting reads BLAST as ‘on target.’ In practice, the distribution of variant scores is bimodal and highly concentrated at 0 and 1, so results are insensitive to the exact threshold (Supplementary Fig. S2).
4.2 Filtering mouse calls from PDX samples
One important use case for MAPEX is as a post-variant-calling filter for PDX samples that have been aligned to a human reference genome. To test the precision of MAPEX, we compared variant calls from aligning reads to the human reference and filtering with MAPEX to calls from two other methods. The first alternate method is to align reads to a combined human and mouse reference and then call variants (Bruna et al., 2016), which we refer to as the ‘combined reference’ method. This requires similar CPU time to using MAPEX. The second method is to align reads separately to human and mouse references and call variants using only those reads that align better to the human reference, which is the method implemented in bamcmp (Khandelwal et al., 2017). This requires twice as much CPU time for alignment as MAPEX, and the post-alignment step is typically shorter for MAPEX, although it can be longer for samples with very high mouse contamination (Supplementary Fig. S3). For three representative PDX tumors, all three methods yield similar callsets (Fig. 2A). The differences are primarily confined to low-frequency variants, and almost all high-frequency variants are called by all three methods (Fig. 2B). MAPEX might reduce power to identify low-frequency subclonal variants, if some of the few reads supporting a variant BLASTed to incorrect locations. This would yield an intermediate variant score. Because variant scores are strongly bimodal (Supplementary Fig. S2), we expect that MAPEX causes little to no reduction in power. Across 34 PDX tumors, all three methods yield a similar dramatic reduction in called variants (Fig. 2C).
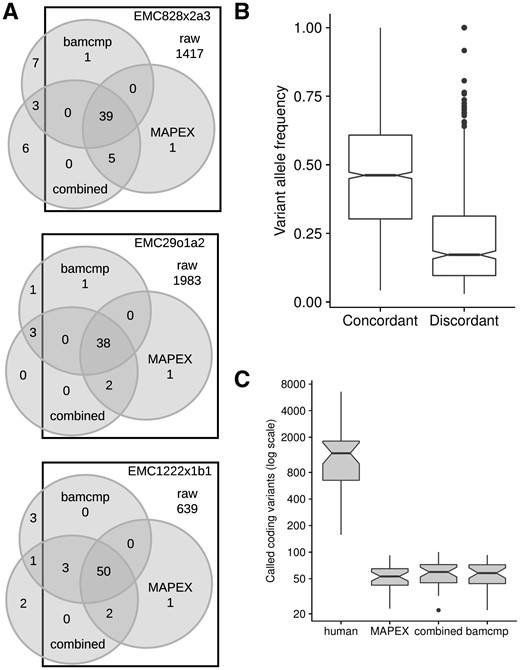
Comparison of MuTect 1.1.1 variants calls between MAPEX, combined reference, and bamcmp methods. (A) Detailed breakdown of variant call overlap between the unfiltered human alignment (squares), MAPEX filtered human alignment (right circles), bamcmp filtered human alignment (top circles) and unfiltered combined alignment (bottom circles) for representative PDXs created from three different primary tumors. (B) Variant allele frequencies for calls in 34 PDX samples that are concordant (n = 1663 variants) and discordant (n = 552 variants) between the methods. (C) Comparison of total calls between the methods, n = 34 PDX samples. Boxplots depict 25th and 75th percentile with 1.5 IQR whiskers. Notches are Median 1.58 IQR/sqrt(n), and represent rough estimates of 95% confidence interval around the median
To further validate MAPEX, we compared PDX variant calls before and after filtering to the primary tumor from which the PDX was derived, where mouse contamination is not an issue. Across 34 PDX tumors derived from 9 primaries, MAPEX dramatically enriches PDX calls for variants that were also found in the primary tumor and removes few PDX calls that were found in the primary tumor. Among variants in the PDXs, only 0.3% to 10% called before MAPEX filtering were also found in the primary tumor, but 23% to 90% of variants called after MAPEX filtering were found in the primary tumor (Supplementary Table S1). This suggests that MAPEX enriches strongly for true variants. Among variants found both in the primary after MAPEX filtering and in the PDX before MAPEX filtering, 97% to 100% were retained in the PDX after MAPEX filtering (Supplementary Table S1). Only one variant identified in each of two primary tumors was filtered by mapexr in a derived PDX. In primary tumor EMC1222, only 60% (slightly above the 50% cutoff) of variant reads mapped on-target for the primary variant (suggesting that it may be a spurious variant caused by a paralogous sequence), while in the PDXs only 20–45% (slightly below the cutoff) of variant reads mapped on-target. In EMC226, the variant appears to be from human wild-type to mouse wild-type, so 55% of variant reads (in a PDX with 57% mouse contamination), mapped to the mouse genome. Together these results suggest that MAPEX removes few true variants.
To validate the usefulness of MAPEX in practice, we focused on calls within known cancer-associated genes, using the COSMIC database. Among the PDAC samples in COSMIC, 34 genes are mutated in more than 3% of samples. Before filtering with MAPEX, 910 variants were found in these genes among the 34 PDXs we studied. After filtering with MAPEX, only 70 variants were retained. These results suggest that MAPEX removes many false positives, dramatically simplifying variant interpretation. Of particular interest are KRAS, TP53 and SMAD4, which are the most commonly mutated genes in PDAC (Table 1). All of the KRAS mutations filtered by MAPEX are I187V mutants, which result from aligning wild-type mouse KRAS reads to human KRAS, and all 34 PDXs retained the KRAS mutation found in their primary tumor. All of the SMAD4 and TP53 mutations that were retained by MAPEX in the PDXs also appeared in the corresponding primary tumors, and all of those filtered were not found in the corresponding primary tumors. ARID1A is particularly susceptible to spurious variants caused by mouse contamination; only one of the 131 variants originally called in ARID1A was retained by MAPEX. We confirmed that the single retained variant was found in the primary tumor from which the PDX was derived, while none of the 130 rejected variants were found in their corresponding primaries.
Variants detected in PDX samples for important PDAC genes
| Gene | before MAPEX | after MAPEX | ||
|---|---|---|---|---|
| Total | Samples with | Total | COSMIC | |
| variants | a variant | variants | prevalence | |
| KRAS | 56 | 34 | 34 | 0.64 |
| TP53 | 9 | 9 | 7 | 0.39 |
| SMAD4 | 5 | 5 | 5 | 0.14 |
| SYNE1 | 3 | 3 | 0 | 0.05 |
| CSMD3 | 96 | 25 | 0 | 0.05 |
| GNAS | 6 | 6 | 6 | 0.05 |
| HMCN1 | 10 | 5 | 0 | 0.04 |
| APC | 12 | 11 | 0 | 0.04 |
| NEB | 31 | 17 | 0 | 0.04 |
| WDFY4 | 6 | 4 | 1 | 0.04 |
| LRP1B | 32 | 18 | 1 | 0.04 |
| ARID1A | 131 | 33 | 1 | 0.04 |
| Gene | before MAPEX | after MAPEX | ||
|---|---|---|---|---|
| Total | Samples with | Total | COSMIC | |
| variants | a variant | variants | prevalence | |
| KRAS | 56 | 34 | 34 | 0.64 |
| TP53 | 9 | 9 | 7 | 0.39 |
| SMAD4 | 5 | 5 | 5 | 0.14 |
| SYNE1 | 3 | 3 | 0 | 0.05 |
| CSMD3 | 96 | 25 | 0 | 0.05 |
| GNAS | 6 | 6 | 6 | 0.05 |
| HMCN1 | 10 | 5 | 0 | 0.04 |
| APC | 12 | 11 | 0 | 0.04 |
| NEB | 31 | 17 | 0 | 0.04 |
| WDFY4 | 6 | 4 | 1 | 0.04 |
| LRP1B | 32 | 18 | 1 | 0.04 |
| ARID1A | 131 | 33 | 1 | 0.04 |
Variants detected in PDX samples for important PDAC genes
| Gene | before MAPEX | after MAPEX | ||
|---|---|---|---|---|
| Total | Samples with | Total | COSMIC | |
| variants | a variant | variants | prevalence | |
| KRAS | 56 | 34 | 34 | 0.64 |
| TP53 | 9 | 9 | 7 | 0.39 |
| SMAD4 | 5 | 5 | 5 | 0.14 |
| SYNE1 | 3 | 3 | 0 | 0.05 |
| CSMD3 | 96 | 25 | 0 | 0.05 |
| GNAS | 6 | 6 | 6 | 0.05 |
| HMCN1 | 10 | 5 | 0 | 0.04 |
| APC | 12 | 11 | 0 | 0.04 |
| NEB | 31 | 17 | 0 | 0.04 |
| WDFY4 | 6 | 4 | 1 | 0.04 |
| LRP1B | 32 | 18 | 1 | 0.04 |
| ARID1A | 131 | 33 | 1 | 0.04 |
| Gene | before MAPEX | after MAPEX | ||
|---|---|---|---|---|
| Total | Samples with | Total | COSMIC | |
| variants | a variant | variants | prevalence | |
| KRAS | 56 | 34 | 34 | 0.64 |
| TP53 | 9 | 9 | 7 | 0.39 |
| SMAD4 | 5 | 5 | 5 | 0.14 |
| SYNE1 | 3 | 3 | 0 | 0.05 |
| CSMD3 | 96 | 25 | 0 | 0.05 |
| GNAS | 6 | 6 | 6 | 0.05 |
| HMCN1 | 10 | 5 | 0 | 0.04 |
| APC | 12 | 11 | 0 | 0.04 |
| NEB | 31 | 17 | 0 | 0.04 |
| WDFY4 | 6 | 4 | 1 | 0.04 |
| LRP1B | 32 | 18 | 1 | 0.04 |
| ARID1A | 131 | 33 | 1 | 0.04 |
4.3 Effects of variant call filters on PDXs
We carried out our primary analyses with the variant caller MuTect 1.1.1, but to test the performance of MAPEX with other variants callers, we also considered MuTect2 and Varscan 2.
If mouse contamination were perfectly filtered, the number of called variants should not depend on the level of mouse contamination. For all three variant callers the number of raw calls was strongly correlated with estimated mouse contamination (Fig. 3A–C), although MuTect2 and Varscan2 did produce substantially fewer calls overall than MuTect 1. After filtering with MAPEX, the numbers of variants called by all three callers was not significantly correlated with the level of mouse contamination (Fig. 3D–F).
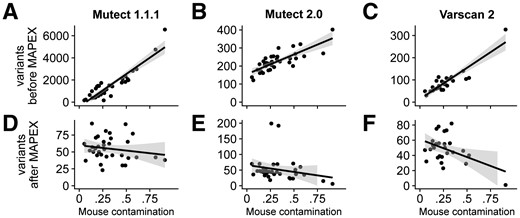
Effects of variant caller on analyzing xenograft samples with MAPEX. (A–C) For all three calling algorithms and 34 xenograft samples (black dots), the number of raw variants called was strongly dependent on estimated mouse contamination. (D–F) After filtering with MAPEX, the number of calls was independent of mouse contamination for all three callers. Lines show linear regressions and shading denotes 95% confidence intervals
Importantly, as a post-variant-calling filter, MAPEX can not evaluate variants that were not initially called. Filters implemented with a variant caller, generally designed to improve results from primary tumors, can cause problems when using MAPEX. For example, MuTect2 applies a clustered event filter designed to reduce the number of false-positive variant calls due to mis-alignment of highly paralogous sequences. In regions of high similarity between mouse and human, this filter can remove true variants. For instance, Figure 4 shows the result of aligning a PDX with modest mouse contamination to the human reference for a small portion of the KRAS oncogene. MuTect 1.1.1 and Varscan 2 both called three variants at this locus, and MAPEX correctly rejected the two spurious variants arising from mouse contamination and retained the true G12D variant. MuTect2 fails to call any of these variants, because they are filtered as likely homologous mapping events, so MAPEX does not see and cannot retain the true G12D variant. In our PDX samples, we found instances of the clustered event filter removing true variants from other PDAC oncogenes, including SMAD4 and TP53.
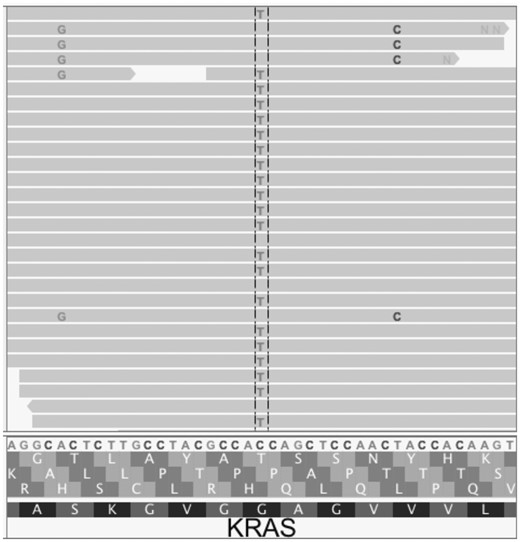
This Integrative Genomics Viewer (Thorvaldsdóttir et al., 2013) window covers a portion of the human KRAS gene. The C > T variant is the classic KRAS G12D mutation that appears in many PDAC tumors. The A > G and T > C variants both result from aligning wild-type mouse reads to the human sequence. When used with MuTect 1.1.1 or Varscan 2, MAPEX correctly retains only the G12D variant. MuTect2, however, filters all three variants, so the G12D variant cannot be retained
Overall, the performance of MAPEX does not depend sensitively on the variant caller used, but callers can introduce specific biases. In particular, the default parameters for Varscan 2 yield high sensitivity but low specificity, so the use of the built-in post-call variant filters is necessary to prevent excessive false positives (Supplementary Fig. S4). In contrast, the default parameters for MuTect2 yield much higher specificity, but at the cost of sensitivity in the PDX context. Currently, the clustered event filter cannot be disabled in MuTect2. We thus advise that users pairing MAPEX with MuTect2 be cautious when interpreting callsets from PDX samples in genes with high similarity between human and mouse.
4.4 Flagging potential false positives resulting from paralogous sequences
In addition to removing mouse contamination from PDX samples, MAPEX can also filter potential paralogs in primary samples. Across 93 PDAC primary tumors, a mean of 11% of total variant calls were flagged by MAPEX as potential paralogs, with a range of 2–33%. The genes in which variants were most frequently flagged as potentially arising from paralogous sequences include members of large gene families, such as mucins, zinc-finger nucleases and the PRAME family (Table 2). Variants in citrate synthase (CS) were also frequently flagged (Table 2). CS has a known pseudogene NCBI: LOC440514 that was responsible for all of the spurious calls. We called variants with MuTect 1.1.1 and filtered with MAPEX, but MuTect2 includes new clustered event and read-mapping quality filters to prevent calling variants caused by paralogs. Using MAPEX yielded call sets that were identical with MuTect2 for all the genes in Table 2, with the exception of MUC12 and MUC5B, which differed by three variants. MAPEX can thus be efficiently and confidently used to remove variants that likely arise from paralogous sequences, with the additional benefit that the reason for classifying a variant as a potential paralog, as well as the genomic locations of the paralogous sequences, can be investigated.
Top genes for which MAPEX flagged variants as potentially arising from paralogs
| Gene | Variants | Samples with |
|---|---|---|
| flagged | a flagged variant | |
| ZNF814 | 15 | 15 |
| CS | 12 | 7 |
| IGFN1 | 8 | 6 |
| KMT2C | 7 | 7 |
| FRG1 | 6 | 6 |
| LILRB3 | 6 | 6 |
| MUC12 | 6 | 6 |
| RGPD3 | 6 | 6 |
| USP6 | 6 | 3 |
| FCGBP | 5 | 4 |
| MUC5B | 5 | 5 |
| NBPF1 | 5 | 3 |
| PRAMEF11 | 5 | 4 |
| PRB4 | 5 | 3 |
| RGPD8 | 5 | 4 |
| Gene | Variants | Samples with |
|---|---|---|
| flagged | a flagged variant | |
| ZNF814 | 15 | 15 |
| CS | 12 | 7 |
| IGFN1 | 8 | 6 |
| KMT2C | 7 | 7 |
| FRG1 | 6 | 6 |
| LILRB3 | 6 | 6 |
| MUC12 | 6 | 6 |
| RGPD3 | 6 | 6 |
| USP6 | 6 | 3 |
| FCGBP | 5 | 4 |
| MUC5B | 5 | 5 |
| NBPF1 | 5 | 3 |
| PRAMEF11 | 5 | 4 |
| PRB4 | 5 | 3 |
| RGPD8 | 5 | 4 |
Top genes for which MAPEX flagged variants as potentially arising from paralogs
| Gene | Variants | Samples with |
|---|---|---|
| flagged | a flagged variant | |
| ZNF814 | 15 | 15 |
| CS | 12 | 7 |
| IGFN1 | 8 | 6 |
| KMT2C | 7 | 7 |
| FRG1 | 6 | 6 |
| LILRB3 | 6 | 6 |
| MUC12 | 6 | 6 |
| RGPD3 | 6 | 6 |
| USP6 | 6 | 3 |
| FCGBP | 5 | 4 |
| MUC5B | 5 | 5 |
| NBPF1 | 5 | 3 |
| PRAMEF11 | 5 | 4 |
| PRB4 | 5 | 3 |
| RGPD8 | 5 | 4 |
| Gene | Variants | Samples with |
|---|---|---|
| flagged | a flagged variant | |
| ZNF814 | 15 | 15 |
| CS | 12 | 7 |
| IGFN1 | 8 | 6 |
| KMT2C | 7 | 7 |
| FRG1 | 6 | 6 |
| LILRB3 | 6 | 6 |
| MUC12 | 6 | 6 |
| RGPD3 | 6 | 6 |
| USP6 | 6 | 3 |
| FCGBP | 5 | 4 |
| MUC5B | 5 | 5 |
| NBPF1 | 5 | 3 |
| PRAMEF11 | 5 | 4 |
| PRB4 | 5 | 3 |
| RGPD8 | 5 | 4 |
5 Conclusion
Genome sequencing is an increasingly important tool in cancer research, but spurious variant calls remain a challenge. MAPEX is an algorithm designed to filter spurious variants caused by mouse reads in PDXs and caused by paralogous sequences in primary tumors. We showed that MAPEX is as sensitive and specific as more computationally intensive methods for calling variants from PDX tumors. We also showed that MAPEX successfully flags variant calls in potentially problematic gene families in primary tumors. Our implementation, mapexr, fits cleanly into standard tumor variant-calling pipelines and runs quickly on modern desktop computers. MAPEX is thus a potentially useful new component for many tumor variant-calling pipelines.
Funding
This work was supported by the National Science Foundation via Graduate Research Fellowship DGE-1143953 to BKM and by the National Institutes of Health via grants R01CA211878-01 and P30CA023074-36S2 to AKW and ESK.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}