





Abstract
Elucidation of protein native states from amino acid sequences is a primary computational challenge. Modern computational and experimental methodologies, such as molecular coevolution and chemical cross-linking mass-spectrometry allowed protein structural characterization to previously intangible systems. Despite several independent successful examples, data from these distinct methodologies have not been systematically studied in conjunction. One challenge of structural inference using coevolution is that it is limited to sequence fragments within a conserved and unique domain for which sufficient sequence datasets are available. Therefore, coupling coevolutionary data with complimentary distance constraints from orthogonal sources can provide additional precision to structure prediction methodologies.
In this work, we present a methodology to combine residue interaction data obtained from coevolutionary information and cross-linking/mass spectrometry distance constraints in order to identify functional states of proteins. Using a combination of structure-based models (SBMs) with optimized Gaussian-like potentials, secondary structure estimation and simulated annealing molecular dynamics, we provide an automated methodology to integrate constraint data from diverse sources in order to elucidate the native conformation of full protein systems with distinct complexity and structural topologies. We show that cross-linking mass spectrometry constraints improve the structure predictions obtained from SBMs and coevolution signals, and that the constraints obtained by each method have a useful degree of complementarity that promotes enhanced fold estimates.
Scripts and procedures to implement the methodology presented herein are available at https://github.com/mcubeg/DCAXL.
Supplementary data are available at Bioinformatics online.
1 Introduction
Elucidation of the three-dimensional functional conformation of proteins is a key step to understand fundamental biochemical processes of living organisms (Alberts, 1998; Alberts et al., 2014; Piccolino, 2000). Although the native state of a protein is directly determined by its amino acid sequence (as stated by Anfinsen's dogma), the very large number of degrees of freedom and consideration of various physico–chemical environments turns the prediction of protein 3-D structures a perplexing problem (Anfinsen, 1973; Dobson et al., 1998; Dobson, 2003; Dill and MacCallum, 2012). During the last two decades, numerous methodologies have been developed to perform in silico prediction of native states of proteins (Baker, 2014; Baker and Sali, 2001; Cooper et al., 2010; Honig, 1999; Rohl et al., 2004; Roy et al., 2010; Webster, 2000;). Although several methods have shown substantial accuracy in identifying folding architectures of specific systems, their applicability is usually limited to comparative modeling or requires massive computational power (Bender et al., 2016; Dill and MacCallum, 2012; Freddolino et al., 2010; Kelley et al., 2015; Piana et al., 2014; Roche and McGuffin, 2016; Yang et al., 2015).
In this context, coevolutionary signals have been used with remarkable success in the identification of inter- and intramolecular protein interactions related to a broad range of functional states (Göbel et al., 1994; de Juan et al., 2013; Morcos et al., 2011; 2014; Taylor et al., 2013). It is based on the principle that during the differentiation of a protein family along divergent evolution, mutation events in residues that are critical to protein functionality are compensated by complementarity mutations (Göbel et al., 1994; de Juan et al., 2013; Morcos et al., 2011; Shindyalov et al., 1994). When sufficient sequence data are available, statistical methods can be applied in multiple sequence alignments (MSAs) to estimate the correlation between these pairwise mutations and to identify co-evolving residues that typically are a proxy for spatial proximity in the native state (Morcos et al., 2011). Several molecular modeling techniques have been successfully adapted to include such co-evolutionary couplings as parameters to assist fold recognition and elucidate the organization of oligomeric complexes (Hopf et al., 2012; Marks et al., 2012; Morcos et al., 2011; Ovchinnikov et al., 2014; dos Santos et al., 2015; Sułkowska et al., 2012; Sutto et al., 2015). RNA structure elucidation has also benefit from the use of global methods to extract residue interactions (De Leonardis et al., 2015; Taylor and Hamilton, 2017; Weinreb et al., 2016).
Another distinct and promising state-of-the-art methodology to infer structural information about biomolecular systems is the combination of chemical cross-linking (XL) and mass spectrometry (MS) techniques (XL-MS) (Sinz et al., 2015; Young et al., 2000). Most commonly, cross-links are obtained by the exposure of a target protein to bifunctional chemical linkers able to react with specific protein residue side chains. Typically, proteins are subjected to tryptic digestion followed by MS analysis. Identification of modified peptides provides information from pairwise maximum distance limits that can be used to restrict search through the protein conformational space (Sinz, 2006; Sinz et al., 2015). Recent advances in mass spectrometry instrumentation, the establishment of robust cross-linking protocols and the development of specialized software for cross-linking identification have expanded the applicability of XL-MS to assist protein fold determination and complex predictions (Brodie et al., 2017; Hofmann et al., 2015; Jin Lee, 2008; Liu et al., 2015; Nguyen-Huynh et al., 2015; Paramelle et al., 2013; Petrotchenko et al., 2014; Pereira et al., 2014; Santos et al., 2011; Sinz, 2006 ).
Concerning the conformational search intrinsic to any structural prediction method, the use of simplified representations of the protein structure allows an efficient search of the conformational space. For example, coarse-grained models using only Cα atoms are proven to be practical in the context of structure prediction from coevolutionary constraints and also for the analysis of folding energy landscapes (Bryngelson et al., 1995; Onuchic et al., 1997; Onuchic and Wolynes, 2004; Wolynes et al., 1995). These simplified models, which originally were conceived using distance constraints obtained from the crystallographic models are called structure-based models (SBMs) and proved to properly represent not only native states but also the multi-dimensional energy funnel that allows the observation of ensembles of intermediate states (Bryngelson et al., 1995; Dill et al., 2008; Onuchic et al., 1997; Wolynes et al., 1995). With an SBM, it is possible to efficiently explore the energy landscape of folding implied by structural data, which is incorporated as interaction potentials in biased molecular dynamic simulations (Clementi et al., 2000; Noel et al., 2010; Onuchic and Wolynes, 2004; Whitford et al., 2009). Lately, the integration of SBM and coevolutionary signals has shown to be an efficient framework to study the protein conformational changes (Morcos et al., 2013; Sfriso et al., 2016), complex formation (dos Santos et al., 2015) and the functional conformation of small globular systems (Sułkowska et al., 2012). Recent coarse grained models like Associative Memory, Water Mediated, Structure and Energy Model (AWSEM, Chen et al., 2016; Davtyan et al., 2012) that include memory terms for fragments and optimized potential have also integrated evolutionary restrains successfully to make estimates of protein folds (Sirovetz et al., 2017).
In this study, we show that SBMs can be used to obtain structural models of the tertiary structure of proteins by incorporating distance constraints obtained from coevolutionary information with those obtained by chemical cross-linking mass-spectrometry, in an efficient and complementary fashion that leads to more robust and accurate structural predictions.
2 Materials and methods
2.1 Estimation of coevolutionary couplings
The comparison of protein sequences within a specific domain can provide information about correlated mutations and aid the inference of physical contacts among residues (Göbel et al., 1994; Shindyalov et al., 1994). A very effective method to compute direct couplings in a MSA, that are typically predictors of physically interacting residue pairs, is direct coupling analysis (DCA; Morcos et al., 2011; Weigt et al., 2009). A detailed description is provided in Supplementary Section S1.
We have used coevolutionary information from DCA to predict protein structures in the past for several systems (Sułkowska et al., 2012), an approach also used successfully by several others (Hayat et al., 2015; Hopf et al., 2015, 2012; Michel et al., 2017; Ovchinnikov et al., 2017). In this work, we are concerned about the effects of integrating experimental data with such coevolutionary signals to improve the process of structural estimation. For this purpose, we selected a set of five protein systems (Table 1) with different degrees of evolutionary coupling accuracies (Cherfils et al., 1997; Luhavaya et al., 2015; Ohren et al., 2007; Stenkamp, 2008; Trajtenberg et al., 2014; Zhang et al., 2010). For all these systems, MSAs were obtained from the Pfam protein family database. Top DCA pairs with highest correlation were selected in equal number L to the length of domain in MSAs. A number of couplings close to the length of the protein have been proposed to be sufficient for efficient structure determination (Kamisetty et al., 2013).
Systems selected for folding prediction
| System | PDB | Pfam ID | sequences | I ± SD (%) | DL | FL |
|---|---|---|---|---|---|---|
| SalBIII | 5CXO | PF12680 | 8806 | 0.17 ± 0.05 | 104 | 134 |
| DesR | 4LE1 | PF00072 | 31596 | 0.25 ± 0.07 | 111 | 132 |
| RAP2A | 1KAO | PF00071 | 16898 | 0.3 ± 0.1 | 160 | 167 |
| Rhodopsin | 3C9L | PF00001 | 27067 | 0.20 ± 0.07 | 252 | 326 |
| Abl Kinase | 3K5V | PF07714 | 16405 | 0.30 ± 0.08 | 250 | 286 |
| Creatine Kinase | 1U6R | PF00217 | 1182 | 0.3 ± 0.2 | 214 | 380 |
| PF02807 | 778 | 0.5 ± 0.2 | 71 |
| System | PDB | Pfam ID | sequences | I ± SD (%) | DL | FL |
|---|---|---|---|---|---|---|
| SalBIII | 5CXO | PF12680 | 8806 | 0.17 ± 0.05 | 104 | 134 |
| DesR | 4LE1 | PF00072 | 31596 | 0.25 ± 0.07 | 111 | 132 |
| RAP2A | 1KAO | PF00071 | 16898 | 0.3 ± 0.1 | 160 | 167 |
| Rhodopsin | 3C9L | PF00001 | 27067 | 0.20 ± 0.07 | 252 | 326 |
| Abl Kinase | 3K5V | PF07714 | 16405 | 0.30 ± 0.08 | 250 | 286 |
| Creatine Kinase | 1U6R | PF00217 | 1182 | 0.3 ± 0.2 | 214 | 380 |
| PF02807 | 778 | 0.5 ± 0.2 | 71 |
Note. DL: pfam domain length; FL: full protein length; I: mean identity of multiple alignment.
Systems selected for folding prediction
| System | PDB | Pfam ID | sequences | I ± SD (%) | DL | FL |
|---|---|---|---|---|---|---|
| SalBIII | 5CXO | PF12680 | 8806 | 0.17 ± 0.05 | 104 | 134 |
| DesR | 4LE1 | PF00072 | 31596 | 0.25 ± 0.07 | 111 | 132 |
| RAP2A | 1KAO | PF00071 | 16898 | 0.3 ± 0.1 | 160 | 167 |
| Rhodopsin | 3C9L | PF00001 | 27067 | 0.20 ± 0.07 | 252 | 326 |
| Abl Kinase | 3K5V | PF07714 | 16405 | 0.30 ± 0.08 | 250 | 286 |
| Creatine Kinase | 1U6R | PF00217 | 1182 | 0.3 ± 0.2 | 214 | 380 |
| PF02807 | 778 | 0.5 ± 0.2 | 71 |
| System | PDB | Pfam ID | sequences | I ± SD (%) | DL | FL |
|---|---|---|---|---|---|---|
| SalBIII | 5CXO | PF12680 | 8806 | 0.17 ± 0.05 | 104 | 134 |
| DesR | 4LE1 | PF00072 | 31596 | 0.25 ± 0.07 | 111 | 132 |
| RAP2A | 1KAO | PF00071 | 16898 | 0.3 ± 0.1 | 160 | 167 |
| Rhodopsin | 3C9L | PF00001 | 27067 | 0.20 ± 0.07 | 252 | 326 |
| Abl Kinase | 3K5V | PF07714 | 16405 | 0.30 ± 0.08 | 250 | 286 |
| Creatine Kinase | 1U6R | PF00217 | 1182 | 0.3 ± 0.2 | 214 | 380 |
| PF02807 | 778 | 0.5 ± 0.2 | 71 |
Note. DL: pfam domain length; FL: full protein length; I: mean identity of multiple alignment.
2.2 Cross-linking/mass spectrometry constraints
We combined DCA constraints, i.e. couplings with high Direct Information (DI) values, with chemical cross-linking/mass-spectrometry (XL) constraints. The constraints were obtained either experimentally or by modeling the expected constraints from the crystallographic model using the Topolink software (Martinez et al., 2017), which computes the solvent-accessible paths and distances for a linker connecting potentially reactive residues. Effective maximum distances for each type of cross-linking considered are listed in Supplementary Table S2.
The experimental dataset for SalBIII (eXL) was comprised by 38 constraints, which resulted from the use of commonly commercial DSS cross-linker and a novel chemistry, named Xplex, which is able to use 1, 6-hexanediamine as a linker as well as to produce simultaneously the formation of zero-length species (unpublished data). No evidence for quaternary cross-links was obtained, suggesting that SalBIII was monomeric in solution. A scheme of the possible linked residues and the experimental constraints distribution over SalBIII sequence is presented in Supplementary Figure S1.
In the case of the SalBIII system, an ideal cross-linking experiment would provide 74 constraints, as predicted by Topolink. Experimentally, 38 constraints were recovered (51%). This relation was used to estimate the limitations in XL experimental determination. Therefore, for the other examples, for which experimental restrictions are not available, XL constraints were obtained by randomly selecting 50% of the crosslinks predicted by Topolink, analogous to the observed SalBIII results.
2.3 Estimation of pairwise equilibrium distances for DCA constraints
Previous conformational studies using DCA signals as interaction potentials for SBM systematically employ a unique equilibrium distance for Cα pairs to represent predicted interaction restrictions independently of residue types (dos Santos et al., 2015; Schug et al., 2009; Sfriso et al., 2016). In an effort to provide a better description of residue–residue interaction distances, we performed a statistical analysis of a large dataset of protein conformations from protein data bank (PDB). We analyzed 43 606 deposited crystallographic structures within 2 Å resolution and computed the Cα–Cα distances for all physically interacting pairs in unique chains. An statistical estimator corresponding to the peak of the distance distribution was designated as equilibrium distance for each specific pair of residue types (Supplementary Figure S2). This estimation resulted in a general improvement in prediction accuracy.
2.4 SBMs
In order to explore the folding of a diverse set of proteins driven by coevolution and XL/MS signals, initial unfolded models for each system were generated using coarse-grained SBMs with residues represented only by Cα atoms (Clementi et al., 2000; Matysiak and Clementi, 2004). These unfolded models are composed by a linear arrangement of Cα beads with null parameters for all dihedrals. Parameters for bound interactions were generated using an in-house algorithm (available at: https://github.com/mcubeg/DCAXL). Bonding angles and dihedrals for each region of protein sequence were estimated by computing the secondary structure with Jpred (Drozdetskiy et al., 2015) and setting optimal parameters based on ideal α-helix and β-sheet structures (Supplementary Table S1). Furthermore, this simple strategy allows the application of secondary structure predictions from diverse sources.
Based on the top ranked coevolving pairs from DCA and observed cross-linking, Gaussian-like potentials were generated to represent each pairwise interaction as described in the Supplementary Material (Lammert et al., 2009; Noel and Onuchic, 2012; dos Santos et al., 2015). For cross-linking interactions, a maximum effective cross-linking distance (Supplementary Table S2) was used to approximate a flat harmonic potential by summation of Gaussian functions with distinct equilibrium positions (Supplementary Section S2).
A schematic of the entire process for merging coevolution and cross-linking signals as structure based models is depicted in Figure 1.
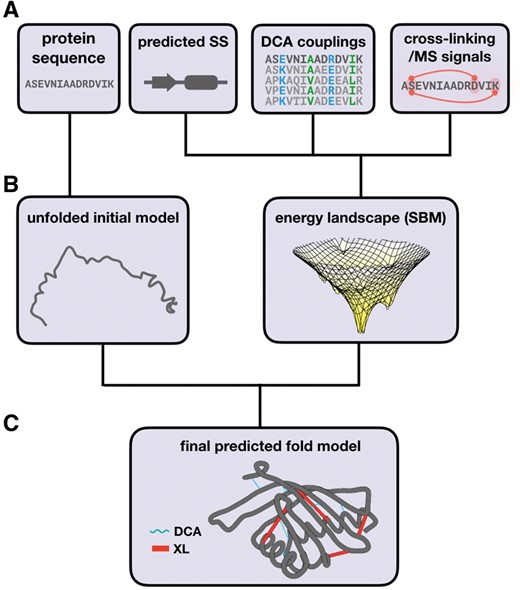
Schematic representation of the steps required for generating protein fold predictions. (A) Primary sequence of a target protein is used to predict the type of secondary structure. An MSA of a protein family is used to estimate coevolving pairs. Interaction signals obtained by chemical cross-linking coupled to MS are also obtained. (B) These datasets are merged to generate an initial unfolded model and a energy landscape (SBM, i.e a customized force-field) for conformational search. (C) Short molecular dynamics simulations using temperature annealing are carried out to identify conformations with optimal restriction agreement
2.5 Folding simulations
Simulations of protein folding were performed using a modified version of Gromacs package with support for Gaussian-like potentials (Noel et al., 2016, 2010; Lammert et al., 2009). Each simulation was developed using an annealing protocol where the system temperature was reduced from 200 to 1 in steps of 100 ps. With this protocol, each folding simulations takes about 1 h of computing time using 4 CPUs (Intel Xeon E5-2670 v2 of 2.50 GHz) for proteins of medium range sizes (250 aa). We performed 1000 independent folding runs for each system to obtain statistically meaningful data on the accuracy of the folding protocol (Fig. 1). Also, the ensemble of models obtained allows for the use of clustering methods for final model evaluation (see Section 2.5). The final template modeling (TM)-score (Xu and Zhang, 2010; Zhang and Skolnick, 2004) and root-mean square deviation (RMSD) values were computed using LovoAlign (Martínez et al., 2007) considering the last frame of each simulation and the reference crystallographic models. All-atom models of folded conformations were constructed with REconstruct atomic MOdel from reduced representation (REMO, Li and Zhang, 2009) from the final Cα coordinates obtained from Molecular Dynamics (MD) simulations with no further refinement.
2.6 Blind selection of correct folds
The modeling performed here generated sets of 1000 models for each target. Therefore, we can explore the properties of the ensemble to classify models, using consensus methods (Kryshtafovych et al., 2014). Here, we opt to use a blind selection of folding conformations from decoy ensembles consisting of evaluating the average similarity of each model to all other models of the ensemble. This classificator is known as the ‘Davis-QA-consensus’ method (Kryshtafovych et al., 2014). All-on-all structural alignments of the models were performed within each ensemble using LovoAlign (Martínez et al., 2007) and the average TM-score was computed for each model.
3 Results
3.1 Coevolutionary and XL/MS signals contribute synergistically to folding prediction
We investigate the contribution of coevolutionary and cross-linking signals on prediction accuracy of the native state of full proteins using SBMs. As an initial case of study, we performed ab initio structure predictions of SalBIII protein from Streptomyces albus and compared with its respective full X-ray structure (PDB ID: 5CXO - chain B), for which experimental cross-linking constraints were obtained recently by Gozzo and co-workers (unpublished data). Coevolutionary constraints were inferred for SalBIII using DCA and the MSA for its family (SnoaL-like domain; Table 1). Top L pairs used for simulations are shown as blue dots in Figure 2. Comparison with monomeric contacts from an X-ray crystal shows substantial agreement of DCA within SalBIII assigned domain. Experimental cross-linking constraints (black circles, Fig. 2) also agree very well with the monomeric X-ray map and are mainly found outside the regions covered by DCA, providing distinct and complementary contact information from that obtained by coevolution. A possible reason to this complementarity is the fact that highly coevolved couplings are originated from interactions that are crucial to preserve minimal function and are usually located in the deep core of macromolecular structure. On the other hand, chemical cross-linking reactions are limited to exposed amino acids and can only account for contacts within surface vicinity. Therefore, important structural information from both sources can be used to get a refined description of interaction patterns.
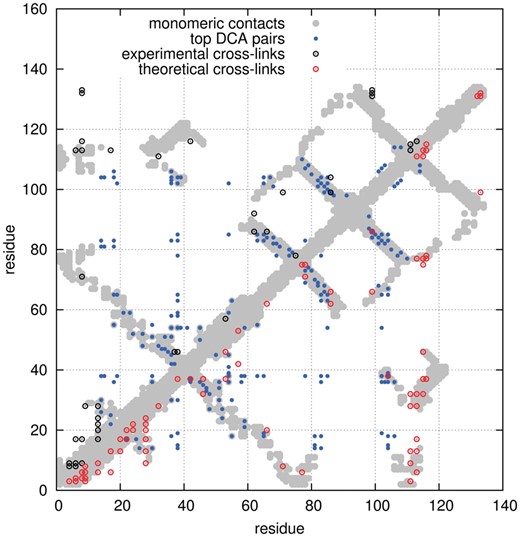
Pairwise interactions used for prediction of SalBIII structure. Comparison of distinct residue–residue distance restrictions obtained from coevolution analysis (blue dots) and experimental and theoretical cross-linking/MS signals (black and red circles, respectively) with monomeric contacts of SalBIII X-ray structure. Physical contacts were computed considering Cα pairs within a distance of 10 Å
In order to evaluate the extent to which cross-linking data can contribute to increase folding accuracy of SalBIII, we also considered an ideal cross-linking experiment corresponding to the set of all possible cross-links that can be expected from the set of linkers used and crystallographic models (red dots, Fig. 2, see Section 2 for details). Consideration of this idealized experiment confirmed the low overlap with evolutionary couplings, evidencing that both techniques can provide unique structural information, which can be utilized to increase prediction accuracy of any computational protocol.
Figure 3 shows the distributions of TM-scores for 1000 simulations in ensembles considering each set of distance restrictions. As expected, simulations using only secondary structure information and no distance restrictions resulted in TM-score values below 0.2 (distributions in grey, Fig. 3 and Table 2), corresponding to random conformations.
Comparison of SalBIII folding prediction using distinct distance constraints
| Restriction | None | DCA | eXL | iXL | DCA + eXL | DCA + iXL |
|---|---|---|---|---|---|---|
| Best TM-score | 0.19 | 0.51 | 0.27 | 0.37 | 0.56 | 0.57 |
| %TM-score > 0.5 | 0 | 1.3 | 0 | 0 | 26.0 | 51.9 |
| Restriction | None | DCA | eXL | iXL | DCA + eXL | DCA + iXL |
|---|---|---|---|---|---|---|
| Best TM-score | 0.19 | 0.51 | 0.27 | 0.37 | 0.56 | 0.57 |
| %TM-score > 0.5 | 0 | 1.3 | 0 | 0 | 26.0 | 51.9 |
Note. DCA: coevolution signals obtained from direct-coupling analysis; eXL: experimental cross-linking/MS signals; iXL: ideal theoretical cross-linking/MS signals based on available X-ray models.
Comparison of SalBIII folding prediction using distinct distance constraints
| Restriction | None | DCA | eXL | iXL | DCA + eXL | DCA + iXL |
|---|---|---|---|---|---|---|
| Best TM-score | 0.19 | 0.51 | 0.27 | 0.37 | 0.56 | 0.57 |
| %TM-score > 0.5 | 0 | 1.3 | 0 | 0 | 26.0 | 51.9 |
| Restriction | None | DCA | eXL | iXL | DCA + eXL | DCA + iXL |
|---|---|---|---|---|---|---|
| Best TM-score | 0.19 | 0.51 | 0.27 | 0.37 | 0.56 | 0.57 |
| %TM-score > 0.5 | 0 | 1.3 | 0 | 0 | 26.0 | 51.9 |
Note. DCA: coevolution signals obtained from direct-coupling analysis; eXL: experimental cross-linking/MS signals; iXL: ideal theoretical cross-linking/MS signals based on available X-ray models.
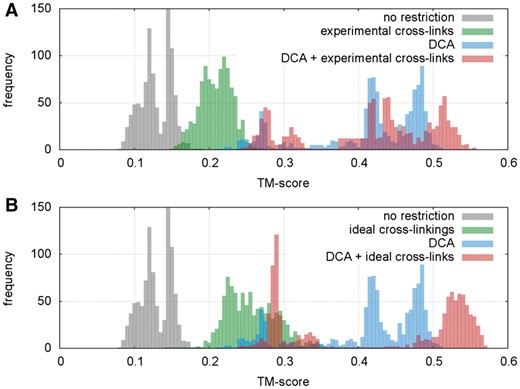
Contributions of distinct distance constraints to folding prediction. Comparison of TM-score distributions for folding of SalBIII protein using coevolution signals and (A) experimental cross-links or (B) theoretical cross-links as interaction data. SalBIII X-ray structure was used as reference model (PDB: 5CXO)
When considering only predicted coevolutionary contacts (Fig. 3, blue distribution) simulations were able to reach folded models with TM-scores relative to the crystallographic model greater than 0.5, meaning that the overall correct fold was obtained (Zhang and Skolnick, 2004), however with low frequency (Table 2). When using exclusively experimental cross-linking signals (38 pairs, see Section 2), an improvement in the TM-score distribution is obtained relative to unrestrained simulations (Fig. 3A, green distributions). Therefore, although the restrictions are quite broad in terms of equilibrium distances, they contribute with meaningful information to structure prediction. Nevertheless, no models with proper folds were obtained.
When considering all possible cross-linking signals (74 pairs), cross-linking constraints provided and additional shift of the ensemble towards higher TM-scores but were still insufficient to achieve fold-level predictions for this system (Fig. 3B, green distributions and Table 2). Therefore, in the context of structure prediction using SBMs, the cross-linking distance restraints appear not to be precise enough to obtain correctly folded structures. This is expected given that neither the SBMs (which in this case do not carry any a priori information on the folded structure) nor the cross-links provide precise residue distance information.
Finally, when we integrate both interactions signals from coevolution and cross-linking, we observe an overall and significant improvement of folding prediction (Table 2). The joint use of experimental cross-links and DCA constraints promoted an increase of 10% in the TM-score of the best predicted model and, most importantly, a 20-fold enhancement of population of models displaying the overall correct fold (Table 2). When considering every possible cross-linking pair along with DCA predictions, we observed an increase of 12% in the TM-score of the best prediction, with an improvement of approximately 40 times on the frequency of simulations reaching the correct fold.
These results motivate the notion that information obtained from coevolution and cross-linking are complementary and can be synergistically applied to increase accuracy and the rate of success in current structure prediction methods.
3.2 Fold of proteins with diverse topologies
We applied the proposed methodology (Fig. 1) to a set of systems with diverse topologies (Table 1). In these cases, cross-linking constraints were determined computationally by using the Topolink package and a random subset of theoretical restrictions (tXL) was utilized to represent the average number of links obtained in XL/MS experiments (see Section 2). Predicted DCA couplings and cross-linking/MS signals for each system are shown in Supplementary Figure S3. A combination of constraints obtained from distinct methodologies (DCA or theoretical cross-linking) promoted substantial improvement in prediction accuracy in all systems selected in this study, when compared with predictions based solely in unique sources of pairwise distance restrictions (only DCA or cross-linking restrictions, Table 3).
Combination of coevolutionary and cross-linking distances restrictions to predict folding of diverse protein systems
| System | Distance restriction | ||||
|---|---|---|---|---|---|
| None | tXL | DCA | DCA + tXL | ||
| DesR | Best TM-score | 0.19 | 0.38 | 0.56 | 0.60 |
| %TM-score > 0.5 | 0 | 0 | 69.7 | 83.5 | |
| RAP2A | Best TM-score | 0.16 | 0.50 | 0.68 | 0.72 |
| %TM-score > 0.5 | 0 | 0 | 80.5 | 75.9 | |
| %TM-score > 0.6 | 0 | 0 | 53.4 | 75.6 | |
| Rhodopsin | Best TM-score | 0.20 | 0.35 | 0.60 | 0.62 |
| %TM-score > 0.5 | 0 | 0 | 79.5 | 81.3 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 12.6 | |
| Abl kinase | Best TM-score | 0.15 | 0.47 | 0.58 | 0.64 |
| %TM-score > 0.5 | 0 | 0 | 58.2 | 58.7 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 40.8 | |
| Creatine kinase | Best TM-score | 0.14 | 0.50 | 0.36 | 0.59 |
| %TM-score > 0.5 | 0 | 0 | 0 | 12.1 | |
| System | Distance restriction | ||||
|---|---|---|---|---|---|
| None | tXL | DCA | DCA + tXL | ||
| DesR | Best TM-score | 0.19 | 0.38 | 0.56 | 0.60 |
| %TM-score > 0.5 | 0 | 0 | 69.7 | 83.5 | |
| RAP2A | Best TM-score | 0.16 | 0.50 | 0.68 | 0.72 |
| %TM-score > 0.5 | 0 | 0 | 80.5 | 75.9 | |
| %TM-score > 0.6 | 0 | 0 | 53.4 | 75.6 | |
| Rhodopsin | Best TM-score | 0.20 | 0.35 | 0.60 | 0.62 |
| %TM-score > 0.5 | 0 | 0 | 79.5 | 81.3 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 12.6 | |
| Abl kinase | Best TM-score | 0.15 | 0.47 | 0.58 | 0.64 |
| %TM-score > 0.5 | 0 | 0 | 58.2 | 58.7 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 40.8 | |
| Creatine kinase | Best TM-score | 0.14 | 0.50 | 0.36 | 0.59 |
| %TM-score > 0.5 | 0 | 0 | 0 | 12.1 | |
Note. DCA: coevolutionary signals obtained from direct-coupling analysis. tXL: theoretical cross-linking/MS signals based on available X-ray models.
Combination of coevolutionary and cross-linking distances restrictions to predict folding of diverse protein systems
| System | Distance restriction | ||||
|---|---|---|---|---|---|
| None | tXL | DCA | DCA + tXL | ||
| DesR | Best TM-score | 0.19 | 0.38 | 0.56 | 0.60 |
| %TM-score > 0.5 | 0 | 0 | 69.7 | 83.5 | |
| RAP2A | Best TM-score | 0.16 | 0.50 | 0.68 | 0.72 |
| %TM-score > 0.5 | 0 | 0 | 80.5 | 75.9 | |
| %TM-score > 0.6 | 0 | 0 | 53.4 | 75.6 | |
| Rhodopsin | Best TM-score | 0.20 | 0.35 | 0.60 | 0.62 |
| %TM-score > 0.5 | 0 | 0 | 79.5 | 81.3 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 12.6 | |
| Abl kinase | Best TM-score | 0.15 | 0.47 | 0.58 | 0.64 |
| %TM-score > 0.5 | 0 | 0 | 58.2 | 58.7 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 40.8 | |
| Creatine kinase | Best TM-score | 0.14 | 0.50 | 0.36 | 0.59 |
| %TM-score > 0.5 | 0 | 0 | 0 | 12.1 | |
| System | Distance restriction | ||||
|---|---|---|---|---|---|
| None | tXL | DCA | DCA + tXL | ||
| DesR | Best TM-score | 0.19 | 0.38 | 0.56 | 0.60 |
| %TM-score > 0.5 | 0 | 0 | 69.7 | 83.5 | |
| RAP2A | Best TM-score | 0.16 | 0.50 | 0.68 | 0.72 |
| %TM-score > 0.5 | 0 | 0 | 80.5 | 75.9 | |
| %TM-score > 0.6 | 0 | 0 | 53.4 | 75.6 | |
| Rhodopsin | Best TM-score | 0.20 | 0.35 | 0.60 | 0.62 |
| %TM-score > 0.5 | 0 | 0 | 79.5 | 81.3 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 12.6 | |
| Abl kinase | Best TM-score | 0.15 | 0.47 | 0.58 | 0.64 |
| %TM-score > 0.5 | 0 | 0 | 58.2 | 58.7 | |
| %TM-score > 0.6 | 0 | 0 | 0 | 40.8 | |
| Creatine kinase | Best TM-score | 0.14 | 0.50 | 0.36 | 0.59 |
| %TM-score > 0.5 | 0 | 0 | 0 | 12.1 | |
Note. DCA: coevolutionary signals obtained from direct-coupling analysis. tXL: theoretical cross-linking/MS signals based on available X-ray models.
Coevolutionary pairs obtained from DCA integrated in SBM potentials showed fold-level accuracy in all systems selected containing single families, with a considerable higher statistics (∼80% for RAP2A and Rhodopsin, Table 3 and Supplementary Fig. S4). Despite cross-link signals only provide upper-limit distances for residue pair interactions, in some cases, the constraints predicted (tXL) were sufficient to drive the simulations towards correct folds (RAP2A, Abl Kinase and Creatine Kinase, Table 3 and Supplementary Fig. S4). These results provide evidence that cross-linking data can improve conformational search and folding predictions and validate this approach as an efficient methodology to assist protein structural characterization.
An special case considered in this study is creatine kinase. This protein contains two distinct conserved domains (Pfam families: PF00217 and PF02807) with limited sequence data, hindering the application of coevolution for structural characterization. As shown in Table 3 and Supplementary Figure S4, using only distance constraints from DCA was insufficient to recover protein native conformations. This same limitation was observed when using only cross-linking restrictions, although a small fraction of predicted conformations achieved near fold-level accuracy (Supplementary Fig. S4). For this case, the combination of DCA and cross-linking signals showed to be crucial to improve the prediction into fold-level accuracy (TM-score > 0.5). This is a representative case of the types of systems where the proposed methodology would be more beneficial.
3.3 Blind selection of native folds
Discrimination of protein native state from folding decoys is a difficult problem (Park et al., 1997). This problem is even more challenging when ab initio predictions provide large decoy ensembles with a plethora of possible folding architectures (Brodie et al., 2017; Cooper et al., 2010; Kosciolek and Jones, 2014; Rohl et al., 2004). Even though there has been a significant refinement improvement in theoretical models describing physical interactions, successful identification of correct folded states based solely in energy functions is rare (Deng et al., 2016; Mishra et al., 2016; Mirny and Shakhnovich, 1996; Sankar et al., 2017; Uziela et al., 2017; Zhou et al., 2014a, 2014b). Recent progress has been achieved using alternative approaches such as entropy estimation and machine learning methods (Sankar et al., 2017; Uziela et al., 2017).
Since we generate an ensemble of 1000 models for each system, we chose to use a consensus method to classify the models (Kryshtafovych et al., 2014). We employed the Davis-QA-consensus classification method for each ensemble of models predicted using DCA and cross-linking/MS data (Table 3). The models with greatest average similarity to all other models in the ensemble were selected. This evaluation allowed to successfully identify the models in the upper limit of TM-score predictions (Fig. 4). Therefore, clustering by similarity resulted to be an effective method for quality assessment of models generated using the current protocol.
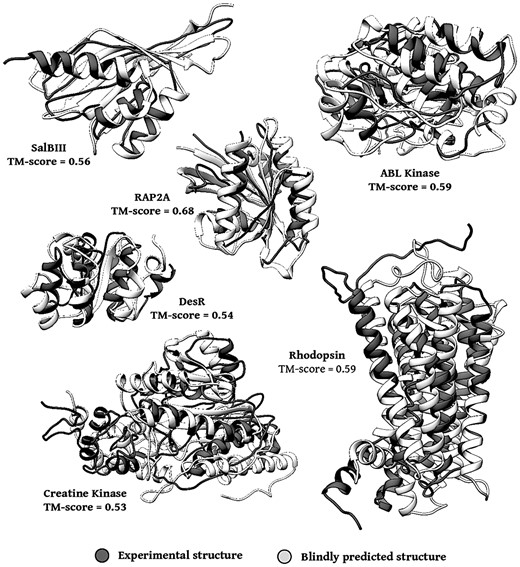
Blind selection of predicted folding using structural similarity analysis. Folded models with TM-scores ≥ 0.5 for all systems considered in this study were identified. Dark gray structures depict blindly selected predictions compared to the experimentally determined structures in light gray. TM-scoreC: predicted model selected by consensus score analysis. TM-scoreB: best model generated, with highest similarity to crystallographic model
4 Discussion
In this work, we provide an effective, computationally inexpensive and robust methodology to predict protein folds using residue–residue couplings from coevolution and cross-linking/MS data integrated with structure based models. We performed a systematic study of the role of each signal component in structure prediction performance for a diverse set of protein topologies. We observe a synergism between coevolutionary and cross-linking restrictions, each contributing with distinct and unique structural information that led to an increase of folding prediction accuracy. While coevolution couplings are usually prevalent in the core of protein structures by key intermolecular contacts that promotes packing, cross-linking reactions are restricted to protein surface due to physical accessibility. Therefore, both components contribute with important information to solve tertiary structure. This is particularly true for the challenging coevolution cases where sequence availability is limited or the domain coverage is insufficient.
Molecular coevolution has recently been established as a significant technique to infer protein interactions and assist structural elucidation. On the other side, the use of experimental cross-linking/MS with long-range linkers as a unique source of interactions is usually insufficient for fine molecular description such as needed for folding prediction (Tamò et al., 2017). Interestingly, although cross-linking signals constitute non-precise distance restrictions, they can provide enough information to allow folding elucidation when substantial data are available (as shown for DesR, RAP2A and Abl and creatine kinases). This observation also suggests that improvement in equipment sensibility for cross-linking/MS signals should boost structural elucidation over the next years.
Finally, we demonstrate how folding ensembles can be used to identify plausible functional conformations by applying a self-consistent similarity analysis. The described methodology can be easily applied to practical problems in structural biology using the protocol and scripts developed in this work and available for others to use. We expect that this approach that integrates and maximizes computational and experimental methodologies will be useful to elucidate important challenges in structural bioinformatics.
Funding
This work was supported by São Paulo Research Foundation [FAPESP, grants: 2015/13667-9, 2010/16947-9, 2013/05475-7, 2013/08293-7; 2016/13195-2; 2014/17264-3] and funds from the School of Natural Sciences and Mathematics at the University of Texas at Dallas.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}