





Abstract
The advent of genomic data from densely sampled bacterial populations has created a need for flexible simulators by which models and hypotheses can be efficiently investigated in the light of empirical observations. Bacmeta provides fast stochastic simulation of neutral evolution within a large collection of interconnected bacterial populations with completely adjustable connectivity network. Stochastic events of mutations, recombinations, insertions/deletions, migrations and micro-epidemics can be simulated in discrete non-overlapping generations with a Wright–Fisher model that operates on explicit sequence data of any desired genome length. Each model component, including locus, bacterial strain, population and ultimately the whole metapopulation, is efficiently simulated using C++ objects and detailed metadata from each level can be acquired. The software can be executed in a cluster environment using simple textual input files, enabling, e.g. large-scale simulations and likelihood-free inference.
Bacmeta is implemented with C++ for Linux, Mac and Windows. It is available at https://bitbucket.org/aleksisipola/bacmeta under the BSD 3-clause license.
Supplementary data are available at Bioinformatics online.
1 Introduction
Simulation models can be used for prediction, parameter estimation and for validating methods used in population genomics (Hoban et al., 2012). Most general-purpose simulators are tailored mainly for eukaryotes (e.g. Arenas and Posada, 2014). However, many studies on evolutionary processes in bacteria have emerged recently, using simulation software tailored for their specific purposes (Fraser et al., 2007; Marttinen et al., 2015; Niehus et al., 2015; Numminen et al., 2016; Teixeira et al., 2017). Simulators can be divided into two categories (Hoban et al., 2012): coalescent simulation starts with the present-day population and simulates backwards in time, coalescing individuals until the most recent common ancestor is found, while forward simulators maintain a population of individuals and simulate forward in time by sampling the next generation from the current one. In general, coalescent simulators are faster, by only considering the ancestors of the current individuals, but forward simulation allows greater flexibility to define the model. This makes the latter attractive for bacteria, where recombination shuffles genetic material between genomes in a complex manner that depends, for example, on the genetic and physical distance between the donor and recipient strains. Furthermore, recombination may cause different parts of the genome to have completely distinct population histories (Feil et al., 2001; Mostowy et al., 2017), undermining the assumption of a single coalescent. The recently published general-purpose simulators tailored for bacteria have all been based on the coalescent approach (Brown et al., 2016; De Maio and Wilson, 2017). Hence, there is a need for an efficient general-purpose forward simulator for bacterial population genomics.
Bacmeta provides an efficient C++ implementation of a finite metapopulation Wright–Fisher model with explicit genome sequences evolving for each strain. Shared C++ pointers and compact object representations result in low memory and run-time requirements. The model allows multiple arbitrarily connected populations, each with thousands of bacteria, for which the genome sequences are subjected to evolutionary events over discrete non-overlapping generations. Bacmeta implements a large variety of different event types governed by user-defined parameters using simple textual input files, which provides a convenient framework for large-scale simulations, integration with other software and likelihood-free inference. For example, Bacmeta could be used for testing methods for inferring recombination (Croucher et al., 2015; Didelot and Wilson, 2015; Mostowy et al., 2017), since every past evolutionary event can be stored to provide the ground-truth, or for likelihood-free inference for model parameters, as in De Maio and Wilson (2017); Marttinen et al. (2015); Numminen et al. (2016); based on the Approximate Bayesian Computation (Beaumont et al., 2002; Lintusaari et al., 2017).
2 Features
The simulation of each evolutionary event, including reproduction, is executed at each generation for a set number of iterations. Possible event types are displayed in Figure 1A and B. Each generation ends in the selection of bacteria for seeding the next generation by random sampling with replacement. The events are modeled as Poisson processes with user-defined rate parameters. For migration and micro-epidemic events, we use the parametrization introduced by Numminen et al. (2016). For mutations and recombinations, we use the same approach as Marttinen et al. (2015), except that mutations are generated under an explicit model with user-defined weights for all nucleotide pairs in ACGT. For insertion/deletion sizes we use the model by Benner et al. (1993), with rate defined in relation to mutations. Note that this also allows for higher-level summaries of the data, for example as integer-labeled alleles for each locus, useful for certain types of analyses. Haplotypes can be flexibly represented as genomic islands of any desired length and number, imitating separate genomic regions or secondary chromosomes. Outputs from the simulator include synthetic DNA sequences, pairwise distance measures and several summaries, e.g. counts of the different events. See Supplementary Material for further overview of usage and model of the software. Full descriptions are given in manuals which are available at bitbucket.org/aleksisipola/bacmeta, under folder ‘manuals’ in the source page.
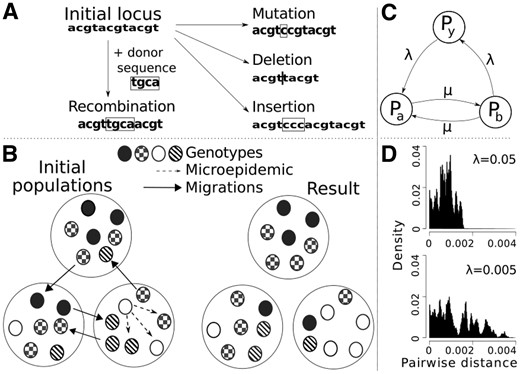
(A) Evolutionary events, (B) population dynamic events, (C) example: the migration connectedness of metapopulation as a network graph, where Py is the observed population and edge weight λ represents the inward and outward migration rates of Py and μ = 0.01. (D) Effect of low versus high value of λ on pairwise distances in population Py
3 Examples
To illustrate the functionality and performance of Bacmeta, we considered the effect of inter-population connectedness via a migration network. We simulated 10 occurrences of 2 scenarios corresponding to low and high connectedness of the observed population, Py, each for 20 000 generations. We used a metapopulation with three populations, with migration routes in Figure 1C, migration rates in Supplementary Table S2, and general parameters in Supplementary Table S1, following values of recombinogenic bacteria. As expected, the higher connectivity led to markedly lower pairwise distances in the observed population Py, reflecting reduced divergence between clusters (Fig. 1D). Run-times were between 30–37 s on a single core of Intel Core i5-7200 U CPU @ 2.50 GHz.
As another example, we considered how distances between populations evolved across one longer simulation in another migration scenario (Supplementary Figs S1 and S2). We found that a complex interaction network may lead to structured populations where tightly linked clusters maintain an equilibrium distance between each other, while loosely linked clusters diverge rapidly. These findings extend previous results considering the special case of two clusters (Marttinen and Hanage, 2017).
Funding
Academy of Finland (COIN Centre of Excellence and grants 286607 and 294015 to PM) and ERC (grant 742158 to JC).
Conflict of Interest: none declared.
References


{kind=link}