





Abstract
In the new release of pymzML (v2.0), we have optimized the speed of this established tool for mass spectrometry data analysis to adapt to increasing amounts of data in mass spectrometry. Thus, we integrated faster libraries for numerical calculations, improved data retrieving algorithms and have optimized the source code. Importantly, to adapt to rapidly growing file sizes, we developed a generalizable compression scheme for very fast random access and applied this concept to mzML files to retrieve spectral data.
pymzML performs at par with established C programs when it comes to processing times. However, it offers the versatility of a scripting language, while adding unprecedented fast random access to compressed files. Additionally, we designed our compression scheme in such a general way that it can be applied to any field where fast random access to large data blocks in compressed files is desired.
pymzML is freely available on https://github.com/pymzML/pymzML under GPL license. pymzML requires Python3.4+ and optionally numpy. Documentation available on http://pymzml.readthedocs.io.
1 Introduction
The rise of high-throughput methods in modern mass spectrometry, has triggered the demand for tools to analyze data with new and complex experimental setups. The two key elements at the forefront of computational mass spectrometry method development are rapid development cycles, by using e.g. scripting languages, such as Python and most importantly standardized file formats.
With pymzML we have pioneered the scripting language access to the mzML file format (Bald et al., 2012), which has been proven to be very powerful in different projects (Kukuczka et al., 2014; Röst et al., 2015). Subsequently, other scripting implementations, such as pyteomics (Goloborodko et al., 2013) and pyOpenMS (Röst et al., 2014), highlighted the need for rapid development cycles. Python is well suited for such a task, due to its high flexibility, extendability and platform independence. Additionally, the Python community is very large, especially in the scientific computing area as manifested by the large number of highly optimized, stable and open source packages. Thus, pymzML v2.0 could be build using the latest optimized libraries for numerical calculations, that e.g. accelerate vectorized calculations (TIC calculation or normalizations). Since pymzML is widely used in computational mass spectrometry method development, we further extended its API towards user written file interface plugins, to allow tailored pipelines for non-standard data analysis to be developed that maintain downstream compatibility. Finally, we integrated the interactive plotting library, plotly, to create a toolbox for visualizing customizable annotated mass spectra and ion chromatograms.
The standard file format for storing and sharing mass spectrometry data is mzML. It was defined by a joint effort of the Seattle Proteome Center/Institute for Systems Biology and the HUPO PSI (Proteomics Standards Initiative) (Deutsch, 2008). mzML is a human-readable XML format, which comes at cost of a large file size due to the high repetition of tags, names and white-spaces. This issue has been addressed by employing compression algorithms, e.g. golomb-rice-coding (Golomb, 1966; Rice and Plaunt, 1971) [aka Numpress (Teleman et al., 2014)] or zlib. These implementations compress the data arrays which constitute most of the data within a mzML file. However, they fail to reduce the size to RAW levels. Currently, the only strategy to achieve such file size reductions is to apply gzip compression in combination with zlib or golomb-rice-coding. Unfortunately, this sacrifices random access the mzML files, which is a major drawback for many computational mass spectrometry approaches.
In this update we enhance the performance of pymzML including highly optimized numerical libraries and we have overcome the limitation in compression approaches by the development of the indexed gzip format (igzip). Our generalized API for igzip allows any data to be stored into a highly compressed gzip file and retrieved by user defined tags, e.g. spectrum IDs in the case of mzML.
2 Materials and methods
We determined general mzML parsing speed in order to evaluate firstly, how the improved pymzML compares to the established C implementation (Fig. 1a) and secondly, how the igzip format alters parsing efficiency in general (Fig. 1b). As a benchmark, we used ten different unindexed mzML files obtained from 4 different mass spectrometers with sizes ranging from 1.3 to 3.8 GB as dataset. Figure 1a shows that C-implementations are still faster [ProteoWizard (Kessner et al., 2008) 2.3×, OpenMS (Röst et al., 2016) 2×] while calculating the total ion current (TIC), i.e. calculating the sum of all intensities in every spectrum, which implies data decoding.
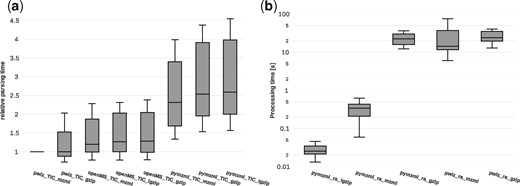
(a) Calculating total ion current (TIC) benchmark comparing OpenMS, Proteowizard and pymzML against each other. (b) Processing times in seconds required for accessing and writing a peak list file from a random spectrum using 10 files varying in size. X-axis, combination of file type and program; Y-axis, processing time in seconds; note log scale
When using gzip compressed files, openMS and Proteowizard perform about 2× faster than pymzML whereas ProteoWizard is slightly faster than openMS. It is important to note, that by tightly integrating numpy into pymzML v2.0, any numerical and vectorial operations on arrays, such as TIC calculations are about 50% faster than the previous pymzML v0.7 (Bald et al., 2012) (data not shown).
On average, uncompressed mzML is 4.9 times larger than RAW and only the combination of zlib or golomb-rice-coding with gzip can reach RAW level sizes (see online documentation). However, files compressed using gzip do not offer random access. While index gzip solutions exist, none are optimized for data blocks that largely exceed the 64KB block size, like in mass spectrometry data. Therefore, we have developed the index gzip format (igzip), which, in combination with golomb-rice-coding or zlib allows files to be compressed to sizes similar or smaller than RAW files while maintaining random access capability (see online documentation for detailed file sizes). Figure 1b shows the random access benchmark, where 10 randomly defined spectra ID were accessed and decoded from each of the benchmark files. In general random access in uncompressed (yet unindexed) mzML is approximately two orders of magnitude faster when using pymzML compared to ProteoWizard’s msaccess command line tool. The fast random access of pymzML v2.0 in uncompressed mzML files is due to our implementation of binary jumps within the file, making the the index information obsolete. Random access in igzip files is faster than in uncompressed files, since a) less data has to be read from disk and b) no binary jumps are used, only one seek operation is required to find the spectrum. A comparison to openMS is not straightforward and was therefore not performed.
3 Conclusion
The tight integration with high performance numerical libraries makes pymzML a very attractive module for computational mass spectrometry.
Additionally, we introduced igzip to enable fast random access in compressed files, allowing mzML files to be compressed back to RAW levels while maintaining random access. Importantly, the igzip strategy can be generally applied to data blocks that largely exceed the 64KB block size, therefore igzip can be used in any other computational context.
Acknowledgements
We thank all contributors over the years for their valuable work with and on pymzML, especially B. Akdeniz, H. Roest and M. Wolfson.
Conflict of Interest: none declared.
References


{kind=link}