





Abstract
The affordability of DNA sequencing has led to the generation of unprecedented volumes of raw sequencing data. These data must be stored, processed and transmitted, which poses significant challenges. To facilitate this effort, we introduce FaStore, a specialized compressor for FASTQ files. FaStore does not use any reference sequences for compression and permits the user to choose from several lossy modes to improve the overall compression ratio, depending on the specific needs.
FaStore in the lossless mode achieves a significant improvement in compression ratio with respect to previously proposed algorithms. We perform an analysis on the effect that the different lossy modes have on variant calling, the most widely used application for clinical decision making, especially important in the era of precision medicine. We show that lossy compression can offer significant compression gains, while preserving the essential genomic information and without affecting the variant calling performance.
FaStore can be downloaded from https://github.com/refresh-bio/FaStore.
Supplementary data are available at Bioinformatics online.
1 Introduction
The growing interest in applications of genome sequencing, together with the dropping costs and continuous improvements in sequencing technologies, has led to the generation of unprecedented volumes of increasingly large and ubiquitous raw genomic datasets (Stephens et al., 2015). These data are characterized by highly distributed acquisition, massive storage requirements, and large distribution bandwidth. For example, the 100 000 Genomes Project has already exceeded 21 Petabytes in size (https://www.genomicsengland.co.uk/the-100000-genomes-project-by-numbers/).
Such flood of data hampers the efficiency of data analysis protocols, limits efficient data sharing and generates vast costs for data storage and IT infrastructure (Schadt et al., 2010). This situation calls for state-of-the-art, efficient compressed representations of the raw genomic data, which cannot only alleviate the storage requirements but also facilitate the exchange and dissemination of these data.
The raw high-throughput sequencing data are primarily stored in FASTQ files (Cock et al., 2010), which are usually considered as the input for the genomic data processing and analysis pipelines. A FASTQ file can be perceived as a collection of ‘reads’, each containing a sequence of nucleotides (generally referred to as the read), the quality score sequence that indicates the reliability of each base in a read, and the identifier, which usually contains the information about the sequencing instrument, flow cell coordinates, and so on. Millions of such reads are produced in a single sequencing run. For example, storing raw whole-genome high-coverage sequencing data of a single human can easily exceed 200 GB in uncompressed form.
Most of the analyses pertaining to DNA sequencing (e.g. in the context of precision medicine) rely on assessing the variants of the sequenced genome against a known reference genome. In order to assess these variants, the raw reads present in the FASTQ file are first aligned to a reference sequence. This process generates aligned reads in Sequence Alignment Map (SAM) format (Li et al., 2009), which contains the same information as the FASTQ file (i.e. the read identifier, the raw sequence of nucleotides and the quality scores), together with the alignment information for each read and possible additional information provided by the mapper.
Although the data contained in the FASTQ file could potentially be recovered from the corresponding SAM file (or its compressed version), there are cases in which this may be impossible. For example: (i) the SAM file may not contain the reads that failed to align to the reference genome or those marked as duplicates, (ii) some reads in the SAM file can be truncated (hard-clipped), (iii) the reference genome used for compression may no longer be accessible during decompression or (iv) there may not be a reference genome at all to generate the corresponding SAM file (e.g. in metagenomics, the different organisms present in the sequenced sample are generally unknown prior to the analysis). Therefore, we focus on the compression of the information stored in FASTQ format, that is, the raw data containing the nucleotide sequences, the read identifiers and the quality scores.
In general, the FASTQ compressors can be divided into four categories: classical, read-mapping-based, read-assembly-based and read-reordering-based. The classical compressors, such as DNA Sequence Reads Compressor (DSRC) (Deorowicz and Grabowski, 2011; Roguski and Deorowicz, 2014), Fqzcomp (Bonfield and Mahoney, 2013) and Quip (Jones et al., 2012), process the reads ‘as they are’, not requiring any additional information nor performing any data preprocessing prior to compression. Although they allow to compress the data while being generated, they achieve moderate compression ratios and the savings in storage space do not scale with the growing sequencing depth. This is in contrast to compressors belonging to the other categories, which focus on efficiently encoding the sequence redundancy of the DNA stream.
For example, read-mapping-based compressors, such as Fastqz (Bonfield and Mahoney, 2013) and LW-FQZip (Zhang et al., 2015)), first map the reads to a provided reference sequence and then encode each read by indicating the location where the read maps in the reference, as well as the mismatches (if any). Read-assembly-based compressors, such as LEON (Benoit et al., 2015) and Quip (Jones et al., 2012), encode the reads in a similar way but using previously built contigs. On the other hand, read-reordering-based compressors, such as Scalce (Hach et al., 2012), ReCoil (Yanovsky, 2011), BEETL (Cox et al., 2012), Overlaping Reads COmpression with Minimizers (ORCOM) (Grabowski et al., 2015) and Mince (Patro and Kingsford, 2015), reorder reads in a manner that ‘similar’ reads are placed close together and hence can be encoded more efficiently. The most promising results, however, seem to be achievable by combining different approaches together, e.g. by read reordering followed by sequence assembly.
Notable examples include recently published proof-of-concept approaches (handling just the DNA symbols, not complete reads) Dynamic Alignment-free and Reference-free Read Compression (DARRC) (Holley et al., 2017) and HAsh-based Read Compressor (HARC) (Chandak et al., 2018), with the latter deserving a special mention. When compressing, HARC hashes all the reads to find the best overlappings between them. Then, it reorders the reads and calculates the consensus sequences (per groups of reads) which are then used as references during the encoding stage. Although it does not handle paired-end reads, it allows to store separately the initial order of the reads. HARC offers a remarkable compression ratio, however, at the cost of needing to keep all the reads in memory along with a number of hash tables, which currently limits its practicality. Moreover, preserving the reads initial ordering (necessary for handling paired-end reads) significantly deteriorates the compression ratio.
The existing specialized solutions for FASTQ files (and DNA-only stream) compression, extensively examined by Numanagić et al. (2016), obtain significant compression gains over general compression tools such as gzip. However, in practice, gzip is still the de facto choice, mainly due to its popularity and stability. It seems that the community has not decided yet that the assets of specialized FASTQ compressors are worth some complications that may appear when moving to a different format of storage.
In this paper we propose FaStore, a new compressor for FASTQ files. FaStore inherits the assets of our previous attempts in the field, especially DSRC (Deorowicz and Grabowski, 2011; Roguski and Deorowicz, 2014), ORCOM (Grabowski et al., 2015) and QVZ (Hernaez et al., 2016; Malysa et al., 2015). The proposed compressor offers both lossless and lossy compression modes and does not use any external reference sequences.
We show that FaStore significantly outperforms the existing compressors in the lossless mode. We, however, advocate for the lossy option when suitable, which, as presented, gives much better shrinkage of the input files with negligible differences in variant calling.
2 Materials and methods
2.1 Overview of the compression algorithm
FaStore is a compressor optimized for handling FASTQ files produced by next-generation sequencing platforms, which are characterized by generating massive amounts of short reads with a relatively low sequencing error rate. FaStore exploits the redundancy present in the reads to boost the compression ratio. In addition, it includes several compression modes to account for the different needs that the users may have. In particular, parts of the data, namely, quality scores and read identifiers, can be optionally discarded or quantized for additional file size reduction. On the other hand, the sequences of nucleotides (DNA sequences) are always losslessly compressed. Due to the different nature of components of reads (DNA sequences, quality scores and identifiers), FaStore uses different specialized compression techniques for each of them (see Supplementary Methods for details). Moreover, as the sequencing data can be generated from a library in a single- or paired-end configuration, FaStore provides different techniques to handle both cases, guaranteeing that the pairing information between the reads is preserved when available. In the following, when clear from the context, the DNA sequences may be also referred to as reads.
Compression of the DNA sequences is done without the use of any external reference sequences. Relying on a reference sequence for compression requires the availability of the same reference at the time of decompression, which may no longer be accessible, thus making the compressed DNA sequences unrecoverable. Hence, this design choice guarantees perfect reconstruction of the sequences.
The reads produced using next-generation sequencing protocols can be thought of as being randomly sampled from across the genome (Firtina and Alkan, 2016) (the input molecule), and thus their initial ordering in the output file carries no information. With this in mind, FaStore reorders the reads to exploit the existing high similarity among the DNA sequences. The reads are clustered in a manner such that reads coming from neighboring positions in the sequenced genome are likely to belong to the same cluster. This key property allows to encode the reads in an efficient manner (see Subsection 2.2 for details).
As a trade-off between the computation time to cluster the reads and the attained compression ratio, FaStore offers two modes of operation, denoted by C0 (fast) and C1 (default). The C0 mode can be perceived as ORCOM compression method extended by a semi-assembly step with some minor improvements. The clustering process in C1 mode, on the other hand, is more involved, leading to better compression ratios. The decompression speed is similar for both modes.
While the DNA sequences can be efficiently compressed due to the redundancy present in the data, the quality scores have proven more difficult to compress (Bonfield and Mahoney, 2013). Part of the reason is that they are inherently noisy and thus characterized by a high entropy. In addition, preserving precise quality scores is often unnecessary (i.e. some distortion is generally acceptable), in that no cost is incurred on the subsequent analyses performed on the data (Ochoa et al., 2016; Yu et al., 2015). Hence, FaStore offers, in addition to lossless compression, various types of lossy compression modes for the quality scores. In particular, FaStore includes Illumina 8-level binning (https://www.illumina.com/documents/products/whitepapers/whitepaper_datacompression.pdf), a custom binary thresholding and an adaptive scheme based on QVZ (Hernaez et al., 2016; Malysa et al., 2015).
Illumina binning mimics the binning of quality scores performed by the latest Illumina sequencing machines by mapping the resolution of quality scores to eight distinct bins (as presented in Illumina technical note in https://www.illumina.com/documents/products/technotes/technote_understanding_quality_scores.pdf, last accessed March 1, 2018). The binary thresholding quantizes the quality scores according to a user-provided threshold qth, setting the quality values below the threshold to qmin and those equal or above to qmax. Finally, QVZ quantizes the quality scores so as to minimize the rate allocation (number of bits per quality score) while satisfying a distortion constraint. To design the appropriate quantizers, QVZ relies on computing the statistics of the quality scores prior to compression. FaStore gathers these statistics while clustering the reads, and thus there is almost no added computational cost. The quantizers are generated after clustering the reads, and one global codebook per dataset is used. For lossless compression, FaStore uses QVZ in lossless mode.
The read identifiers are initially tokenized to make use of the fact that some tokens are constant, some are from a small dictionary and so on. Moreover, since the complete identifiers are usually unnecessary in practice, FaStore also offers a lossy mode for storing them, either by removing the comments (as mappers do by default) or by completely skipping them.
Regarding storing of the pairing information between the reads (when these were generated from a paired-end library), the FASTQ format does not clearly define how the pairing information should be represented. Currently, there are two main ways to convey this information: (i) it is carried in the read identifiers (i.e. a pair of reads share the same identifier) or (ii) it is encoded at the file-level (i.e. the reads reside on the same lines in two FASTQ files or are stored interleaved in a single file). FaStore preserves this information with the sequences, allowing the identifiers to be removed and generating unique ones per pair of reads when decompressing.
2.2 Compression workflow
In FaStore, the compression workflow has been designed as a multi-step process to exploit the high sequence redundancy present in the sequencing data. It consists of (i) reads clustering, (ii) optional reads re-clustering and (iii) reads compression stages. Each stage is further divided into multiple smaller steps. In this section, we provide a general overview of the compression workflow. A detailed description of the methods used to cluster the reads and to compress the DNA sequences, quality scores and read identifiers can be found in the Supplementary Methods. The workflow is depicted in Figure 1 and can be briefly described as follows.
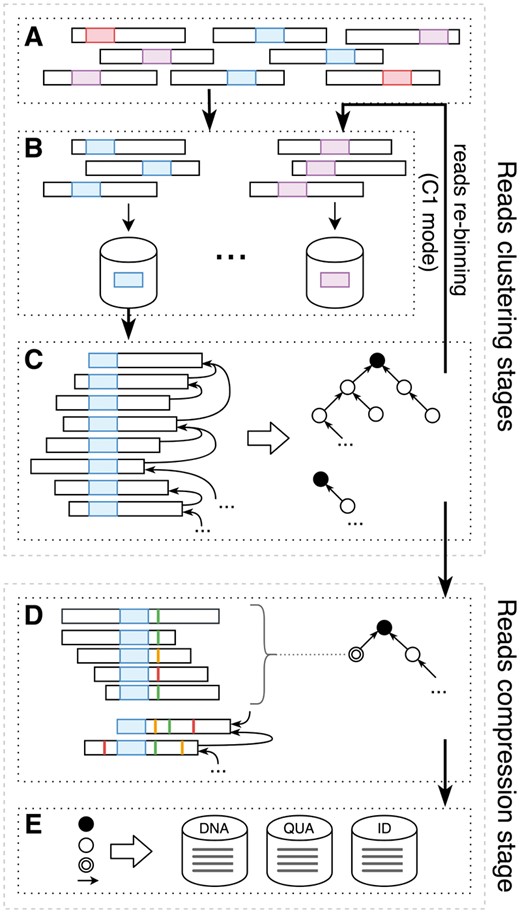
General compression workflow of FaStore. (A) Raw FASTQ reads are (B) distributed into bins according to their signatures (denoted with different colors). (C) Within each bin, the reads are matched, giving as a result a reads similarity graph (black circle represents a node with no matching read found—a root node, while a plain circle represents a standard node). Optionally, the reads follow further re-distribution and matching. (D) With the final similarity graph, the reads are possibly assembled into contigs (denoted with double circle). However, the reads that contain too many mismatching bases with respect to the other ones in the analyzed contig won’t be included in it (the mismatching bases are denoted with colored vertical lines). (E) Finally, all the reads are encoded either in contigs or differentially, depending on the matching result and stored in different streams
The read clustering stage is a two-step process, consisting of a read binning step (see Fig. 1B) and a read matching step (see Fig. 1C). During binning, for each read from the input FASTQ file(s) (Fig. 1A), FaStore seeks the sequence signature, i.e. the lexicographically smallest k-mer, with some restrictions. The signature is used as an identifier of the bin in which the read is placed into. During this step, some statistics are gathered related to the observed DNA sequences, quality scores and read identifiers. At the end of the binning stage, these statistics are used to compute the quantizers for the quality scores, which are stored in a global codebook. This process is performed only when compressing quality scores in QVZ mode. In addition, a token dictionary is built for the read identifiers. These data will be used during the final compression stage.
After binning all the reads, FaStore performs the matching process, independently per each bin. The goal is to find for each read, a referential one, which has the lowest ‘encoding cost’. This cost corresponds to the number of operations required to transform one sequence into another. In order to do so, FaStore first reorders the reads within each bin, so that reads with DNA sequences possibly originating from the same genomic region are likely placed close to each other. Then, we iterate over the reordered reads and, for each sequence, we search in a window of m previous ones for the best match. In general terms, a read can be matched as a normal match, an exact match (an identical sequence was found) or as a hard read (when no satisfactory reference was found). The result of reads matching is represented as a similarity graph, where each node represents a read (DNA sequence) and the edge represents a normal or exact match. More specifically, the result is a forest, where each hard read represents a tree root (a tree can also consist of only a root node). With such graph, we can already proceed to the compression stage (as in C0 mode).
In order to improve the clustering between the sequences, a number of optional reads re-clustering steps can be performed (C1 mode). The goal is to create larger clusters of highly similar (groups of) reads to possibly bring the reads from the same genomic regions close to each other by re-distributing the reads. To do so, we first define a new subset of signatures, which will be used as a filter, to select the bins into which the reads can be moved. Then, for each tree, we select a new root node, which has a new signature residing at the beginning or at the end of its sequence. The connections between nodes are updated and the trees are moved into bins (similarly as in Fig. 1B) denoted by their root signatures, where each tree is represented in the new bin as a single read (its root). This allows to improve the clustering between the reads, by performing an additional matching of them (as in Fig. 1C) and, as a result, building larger trees of similar reads. In the C1 mode, three re-distribution steps are performed.
The compression stage is a two-step process, consisting of assembling the DNA sequences (Fig. 1D) into contigs and encoding the reads (Fig. 1E). First, we traverse each tree and try to assemble the reads into possibly large contigs. The goal is to encode the reads with respect to the built consensus sequences, encoding only the variants (if present) in the contigs. While assembling a contig, for each read, we try to anchor it into the consensus sequence using the position of its signature (which resides at the ‘center’ of the consensus). To add the read to the contig, we assess whether it does not introduce too many variants into the current consensus sequences, as they will need to be encoded by the other reads already present in the contig. When no more reads can be added to the contig, its final consensus sequence is determined by majority voting. As a result, in the graph some of the nodes are replaced with the contig nodes, updating the connections between nodes accordingly.
Finally, we proceed to encode the reads data (Fig. 1E), storing the result in a number of streams, separately for DNA sequences, quality scores and read identifiers. The read sequences are encoded either in contigs (encoding differentially versus consensus sequences) or differentially versus each other, depending on the matching result. To encode the quality scores using QVZ, we use the quantizers from the previously created codebook. Alternatively, when using Illumina 8-level binning or binary thresholding, we encode the transformed quality values. In parallel, we encode the read identifiers using the previously built dictionary. Finally, the streams are compressed using a custom arithmetic coder or the general-purpose compressor PPMd.
3 Results
3.1 Compression factors
For evaluation of the proposed compressor FaStore, we use a subset of datasets already benchmarked by Benoit et al. (2015), Deorowicz and Grabowski (2013), Grabowski et al. (2015) and Numanagić et al. (2016), alongside new ones characterized by a high coverage. The details of the employed datasets are summarized in the Supplementary Methods. The collection consists of seven large sets of paired-end FASTQ files and one vast paired-end dataset, and it includes sequencing data from the Homo sapiens, Gallus gallus and Caenorhabditis elegans species. We compared the performance of FaStore with that of gzip (the de facto current standard in storage of sequencing data) and the top FASTQ compressors according to (Numanagić et al., 2016): DSRC 2 (Roguski and Deorowicz, 2014), Fqzcomp (Bonfield and Mahoney, 2013), Leon (Benoit et al., 2015), Quip (Jones et al., 2012) and Scalce (Hach et al., 2012). We also tested the top DNA-only compressors according to Numanagić et al. (2016): ORCOM (Grabowski et al., 2015), Mince (Patro and Kingsford, 2015), and BEETL (Cox et al., 2012). However, since these algorithms fail to compress the whole FASTQ file, we relegate their results to the Supplementary Worksheet W1. Unfortunately, for several datasets, Mince ran out of available memory (128 GB) and BEETL failed to process some of them in 48 hs time, so they are not included in our analysis.
Figure 2A–D show the average compression factor and (de)compression speeds for the complete collection. All compressors were run using eight processing threads, when applicable, and in maximum compression mode (machine specifications are provided in the Supplementary Material). Due to space constraints and ease of exposition, we provide results for the main lossy settings of FaStore (denoted by reduced, lossy and max) and refer the reader to the Supplementary Worksheet W1 for an extensive evaluation of the whole range of lossy modes provided by FaStore.
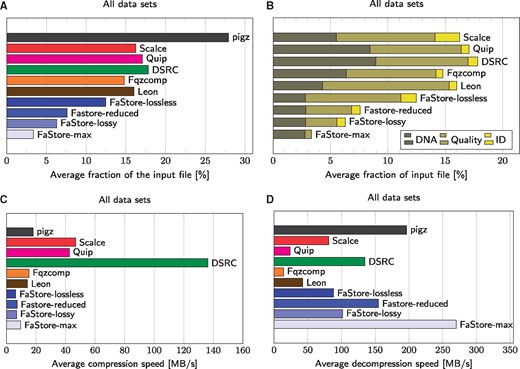
Compression results. (A) Average faction of the input file in % (compressed size divided by original size) for all examined datasets. (B) Average faction of the input file in % for all examined datasets, divided by the different components: DNA bases, quality values, and IDs. (C) Average compression speeds for all examined datasets. (D) Average decompression speeds for all examined datasets. pigz is a multithreaded variant of gzip (same compression ratios, but faster processing)
As shown in Figure 2A–D, FaStore in the lossless mode (preserving all the input data) achieves significantly better compression factors than the competitors. In particular, the compression gains with respect to the results achieved by the best competitor (i.e. Fqzcomp for all datasets except for dataset HSX where Leon outperforms Fqzcomp), range from 7.6% to 20.3%. For example, for H.sapiens datasets HS2 and WGS-42, this corresponds to more than 10 GB of savings in both cases.
Although the lossless mode is used by default in FaStore, we strongly recommend considering some of the provided lossy modes. By discarding parts of the read identifiers and reducing the resolution of the quality scores, one can achieve significant savings in storage space. For example, in the reduced mode, the compressed size is about 60% of that of the lossless mode. The improvement is possible thanks to Illumina 8-level binning and removal of the comments from the identifiers (which, in some cases, leads to storing only a library name and a read number). As the identifiers are usually truncated in this way by mappers when producing SAM files and the eight-level binning becomes a default option in modern sequencers (although the actual mapping of the values can depend on the internal configuration of the sequencing machine), this setting seems to be a reasonable choice.
Even better results (approximately half the size of the lossless mode) are possible when QVZ with distortion level two is applied (lossy mode). Nevertheless, the best compression factor (about a quarter of what was obtained in the lossless mode) is achievable when the identifiers are removed (only the pairing information between the reads is preserved) and the binary thresholding for the quality values is applied (max mode). In our experiments, we selected the values of thresholding parameters ad hoc. We used qth = 20 (as, commonly, values above 20 represent an acceptable quality data, i.e. the probability that the base was incorrectly called is lower than 1 in a 100), qmin = 6 and qmax = 40 (these values were chosen as they represent the lowest and the highest value for a properly called base as in the proposed Illumina bining scheme).
Figure 2B shows fractions of archives consumed by various components: DNA sequences, quality values and read identifiers. There is no result for gzip as, due to the design of this compressor, it is impossible to measure the exact fraction of each component. The results show that FaStore uses much less space to store the DNA sequences than the competitors. Since in the lossy modes there is no loss incurred in the DNA sequences, the amounts of space necessary for storing them are almost identical across the different lossy modes. However, one needs to note that in the lossless mode, FaStore needs more space for storing identifiers than the other competitors (except for Scalce, which also reorders the reads present in the FASTQ files to aid compression). The reason is that after reordering the reads it is much harder to compress their identifiers, as the neighboring ones differ more than in the original ordering. Nevertheless, the compression gain from the sequences of DNA overshadows the compression loss from the identifiers stream.
As outlined earlier, the most difficult to compress are, however, the quality scores. For most compressors, when the quality scores are losslessly compressed, they require more space than the DNA sequences and read identifiers together. Thus, applying the lossy schemes for the quality values has a remarkable impact on the total compression factor. For example, to losslessly compress H.sapiens dataset HS2, FaStore requires 46.3 GB of space. From those, 32.7 GB correspond to the quality scores, which can be further reduced to 9.3 GB (Illumina binning, reduced mode), 8.4 GB (QVZ with distortion level 2, lossy mode) or even 1.1 GB (binary thresholding, max mode). Note that in all the cases, the overall size of the losslessly compressed FASTQ file is reduced by more than 50% when lossy compression of quality values is applied. In particular, a reduction from 46.3GB to as little as 14.7 GB is achieved when binary thresholding is used. Furthermore, this reduction in total size is computed without considering lossy compression of the identifiers, which would provide even more storage savings. In Subsection 3.3, we demonstrate that such reductions in size are possible with little effect on variant calling.
Finally, Figure 2C and D shows the compression and decompression speeds for the different methods analyzed in this paper. The compression speed is some drawback of our solution, as it is somewhat smaller than 10 MB/s (in the default C1 mode). Nevertheless, the decompression speed is comparable to the fastest algorithms, i.e. DSRC 2 and gzip. For use cases where compression speed is of uttermost importance, FaStore provides a fast mode, namely the C0 mode. This mode trades the compression ratio of DNA sequences for compression speed, while still achieving better compression ratios than the competitors (see Supplementary Worksheet W1).
As reported in Supplementary Table S3, when losslessly compressing FASTQ data, on average, the compression speed offered by C0 mode is greater by a factor of 5 as compared to C1 mode at a cost of increasing the size needed to store the DNA bases by a factor of 1.09. Note that switching between C0 and C1 has no significant effect on compression of quality scores and read identifiers, and the speed of decompressing files created in either of these modes is almost identical.
We also evaluated the performance of FaStore on metagenomic datasets. Due to space constraints, the detailed results are provided in the Supplementary Section 5.3. The general conclusions from the experiments are that FaStore offers better compression ratios than the competitors, but the advantage over the best of them, Fqzcomp, is smaller than in the main experiments.
The peak memory and disk usages depend on the dataset size, number of threads and lossy settings. For example, for the largest of our datasets, WGS-42, the RAM consumption ranged from 34 GB (max mode) to 52 GB (lossless mode). These values apply to both the compression and decompression stages. The temporary disk usage (just during compression, as in the decompression we do not use temporary files) varied from 59 GB to 314 GB, respectively. More detailed results are given in Supplementary Table S4.
3.2 Influence of coverage
We also analyzed the compression ratio just for the DNA symbols (Fig. 3) using whole-genome sequencing data of H.sapiens (WGS-235 dataset) sampled at various coverages. As can be noted, for some algorithms (Scalce, Leon, Fqzcomp, ORCOM and FaStore) the increasing coverage leads to significant improvements in compression ratio. In the case of FaStore, the advantage is more than 2-fold over the competitors. When testing read-reordering algorithms (FaStore and Scalce), we also added a series of values in which the reads were compressed as single-end (i.e. the pairing information was lost). For FaStore, this led to further savings in storage space, obtaining about 2.6 times better compression ratios. In addition, FaStore performs consistently better than ORCOM, offering more than 1.2 times improvement in compression ratio and up to 1.3 for the highest coverage data point.
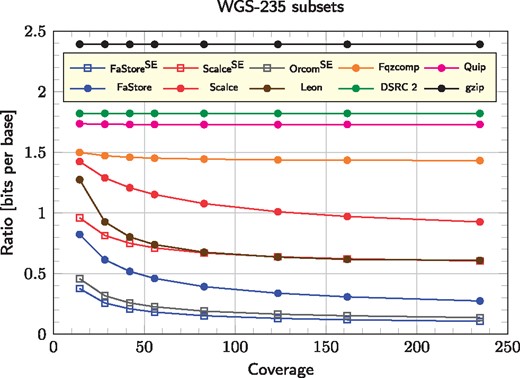
Compression ratio for only DNA symbols (bits to encode a single base) for H.sapiens sampled at various coverages (WGS-235 subsets). Superscript SE stands for single-end
3.3 Variant calling
To investigate the possible side effects of applying lossy compression for base quality scores, we first prepared a set of test FASTQ files, namely, datasets WGS-14 and WGS-42 (see Supplementary Methods), which come from deep sequencing of the NA12878 H.sapiens individual. The reason for this choice is that the National Institute for Standards and Technology (NIST) has released a high-confidence set of variants for that individual (Zook et al., 2014). This allows us to consider this set as the ‘ground truth’ and use it to benchmark the different lossy modes supported by FaStore. The prepared FASTQ files included: (i) original input files (lossless), (ii) original input files with lossy compressed quality scores and (iii) FaStore-shuffled reads with lossy compressed quality scores. Moreover, using WGS-14 dataset, we tested the effect of reordering the reads using an additional set of test FASTQ files. These included (iv) original input files with randomly shuffled reads and (v) FaStore-shuffled reads. A detailed description of the FASTQ files preparation steps can be found in the Supplementary Methods.
With such prepared input FASTQ files, we followed the Genome Analysis Toolkit (GATK) Best Practices recommendations (Auwera et al., 2013) to call the variants (see Supplementary Methods for a detailed explanation of the used pipeline). For assessing the variant calling performance, we used as a ‘gold standard’ the variants for NA12878 provided by the NIST (Zook et al., 2014) and benchmarked our results using the Illumina Haplotype comparison tools pipeline (https://github.com/Illumina/hap.py). In what follows we will report the results on single nucleotide polymorphisms (SNPs), since SNPs are easier to detect and more curated in the high-confidence reference set. Nevertheless, for completeness, results for short insertions and deletions (INDELs) are provided in the Supplementary Worksheet W2. In our experiment, we also focus on the results obtained by applying hard filtering on the called set of SNPs and refer the reader to the Supplementary Worksheet W2 for the complete set of results achieved by applying both hard filtering and semi-automatic filtering Variant Quality Score Recalibration (VQSR) of variants.
Next, we assess the effect that the different lossy quality score compression modes provided by FaStore, namely Illumina binning, binary thresholding and QVZ, have on variant calling. Since QVZ optimizes the quantization for an average mean square error distortion level (specified as an input parameter), for the analysis, we considered distortion levels 1, 2, 4, 8 and 16.
The results of the analysis are presented in Figure 4A and B. We focus on the recall versus precision results obtained when using the considered lossy modes for the WGS-14 and WGS-42 datasets, respectively. It is worth noticing that the precision is similar for both datasets (above 0.99 for most points), whereas the recall is much higher for WGS-42. The majority of points have a recall value around 0.981 for WGS-14 compared to 0.9978 for WGS-42. These results suggest that the sequence coverage plays a key role in variant calling as it improves with increasing coverage.
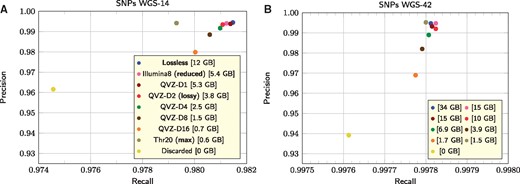
Compression results and variant calling analyses. Results of variant calling for WGS-14 (A) and WGS-42 (B) datasets
The most interesting aspects are, however, the results achieved using the various lossy modes. As expected, increasing significantly the distortion level of QVZ reduces both the recall and the precision. The variant calling performance applying QVZ with distortion level 1 (QVZ-D1) is comparable to that of Illumina binning, with slightly better results (in recall and in precision for WGS-42) in favor of Illumina binning. Moreover, both modes, QVZ-D1 and Illumina binning, offer a similar compression factor, reducing the size of the quality scores by more than 44% when compared to lossless (with slightly better compression achieved by QVZ). In addition, the variant calling performance is almost indistinguishable from that achieved with the original data. Hence, the original size of the WGS-42 dataset could be reduced by an order of magnitude (a 12× reduction), and almost a half of the space required to store it losslessly compressed, by reducing the footprint of quality scores and removing the comments from the read identifiers, all while preserving the variant calling accuracy. As a side note, applying lossy compression to the read identifiers (as done in the reduced and lossy schemes) has no effect on variant calling, as the mappers usually strip the present comments, and can further reduce the compressed size.
In order to further boost the compression gains, more aggressive lossy modes need to be considered at the cost of possibly losing some variant calling accuracy. In that regard, QVZ applied with distortion level 2 (QVZ-D2) seems to be a good trade-off between variant calling performance and compression factor. It offers comparable performance to that obtained with the original data while reducing the size of the quality scores by more than 66%. Distortion levels above 2, although offer significant compression gains, show a degradation on variant calling. For example, with the maximum distortion considered (D16), the precision drops from above 0.99 (with the original data) to 0.98 (WGS-14) and 0.97 (WGS-42). The differences in recall are less pronounced.
Quite surprisingly, for WGS-42 the results for the max mode are almost as good as for the lossless mode (for WGS-14 the recall decreased by about 0.002), with a vast difference on the size of the compressed quality data (0.6 GB versus 12 GB and 1.5 GB versus 34 GB, for WGS-14 and WGS-42, respectively). These results suggest that for datasets with increasing coverage, storing only the information of whether the called base is ‘good’ or ‘bad’ is sufficient for obtaining reliable results on variant calling, while significantly boosting the compression ratio.
For comparison, we also experimented with completely removing the quality data, but the results (series denoted as discarded) show a significant drop in both recall and precision. This indicates that some information about the base quality is necessary for reliable variant calling results (at least in the examined range of coverages).
As mentioned earlier, the reads produced using next-generation sequencing protocols are randomly sampled from across the genome, and thus the original order of the reads carries no meaningful information. Due to the large size nature of the produced data, several commonly used computational methods that operate on these files rely on heuristics to be able to run in a reasonable time (even when executed in multi-threaded mode). For this reason, the reordering of reads, even if theoretically not relevant, may have some effect on variant calling. For example, Firtina and Alkan (2016) showed that, for some mappers, randomly shuffling the input FASTQ reads can lead to different alignment results, especially for reads originating from highly repetitive genomic regions.
Since FaStore permutes the input collection of reads, we examined the impact that various read reorderings can have on variant calling. The goal was thus to analyze how the FaStore-specific shuffling of the reads may affect variant calling. To that end, we compared it against a random shuffling of the reads in the file and against the original order. We included into the comparison the results from different computational environments (differing mainly by the number of computing threads employed).
Figure 1 from Supplementary Methods summarizes the findings of our study for dataset WGS-14. In particular, we compare the Precision and Recall (for the SNPs) with and without applying VQSR filtering, obtained with the original order, the FaStore order and four random shuffles. For the original and FaStore orders, we run the experiments in two different computational environments (hence the two points with identical labels). There are several things to notice from this plot. First, the difference in precision/recall obtained by the various orderings is negligible. More interestingly, this difference is comparable to that of running the experiments in different computational environments, suggesting that the order has no more effect than the used computational environment. For example, when VQSR is not applied, the change in precision is <0.00006, and the change in recall is <0.002. On the other hand, applying VQSR increases the change in precision to 0.02, while maintaining the same variability in recall. More importantly, the choice of applying or not the VQSR filtering has significantly more effect (especially in precision) than the ordering of the reads. The precision drops from 0.98 when no VQSR is applied to 0.91 when applied. This is several orders of magnitude larger than the change due to reordering of the reads. All this put together indicates that the gains in compression obtained by reordering the reads are worth pursuing, as there seems to be little—if none—effect on variant calling.
4 Discussion
The efficient storage and transfer of huge files containing raw sequencing data have become a real challenge. The popular general-purpose compressor gzip is still being used as the de facto standard solution to compress FASTQ files, being able to reduce file sizes by about three times, with significant gains in cost of storage and speed of transfer. However, as already pointed by multiple researchers (Numanagić et al., 2016; Roguski and Ribeca, 2016), even just by splitting the content of the FASTQ file into separate information streams of homogeneous data type (i.e. DNA sequences, read identifiers and quality scores) and compressing each separately using gzip, one can achieve gains in compression of more than 15% than by using gzip alone. Unfortunately, this clearly demonstrates that the current de facto standard approach for storing raw sequencing data is not the best choice and in modern times much higher savings in storage are possible and necessary.
Our proposed compressor, FaStore, is designed to achieve excellent compression factors, i.e. about three times better than gzip and significantly better than the existing specialized FASTQ compressors. In addition, FaStore offers several lossy compression modes for the quality scores and the read identifiers, which result in significant compression gains. Moreover, since the methods to compress DNA sequences, quality scores and read identifiers are modular, they can be possibly implemented in other solutions to store sequencing data in a more compact form. For example, the DNA sequences compression methods can be used in solutions working with SAM alignments to improve the compression of unaligned reads. The methods to compress quality scores can also be applied when compressing alignments in SAM format. Finally, all the methods can be used in different genomic data processing and compression frameworks, e.g. implemented as codecs in CARGO (Roguski and Ribeca, 2016) or Goby framework (Campagne et al., 2013).
In parallel, we strongly suggest the community to consider resignation from storage of all the raw sequenced data or, at least, considering the raw data to be stored in one of the lossy forms. As we presented, together with the increasing sequencing throughput and the dropping costs of sequencing reflected in higher coverages for the smaller prices, the high resolution of quality values seems to be unnecessary. An important stage in this direction was made by Illumina, which has already introduced the possibility of reducing the resolution of the available quality scores to 8 values in some of their newest sequencers (e.g. HiSeq X), and it is considering a more aggressive four-level binning scheme for their latest NovaSeq system. We show that similar variant calling results could be obtained when even more reduction of the quality stream is applied. For sufficiently large coverage it seems to be enough to provide just a binary information about each base telling whether it is ‘good’ or ‘bad’.
To imagine the possible gains in reduction of cost thanks to the lossy approaches let us say that the FASTQ files for H.sapiens sequenced at 42-fold coverage in the paired-end mode could consume as little as 10 GB (FaStore-max), which can be compared to 110 GB of gzipped FASTQ files. For both datasets, the quality of variant calling results should be almost identical.
Acknowledgements
We would like to thank Ivo Gut for supporting the project and Marcos Fernández-Callejo for helpful discussions and technical insights.
Funding
This work was supported by National Science Centre, Poland [under project DEC-2016/21/B/ST6/02153 to S.D.]; European Union Seventh Framework Programme (FP7/2007-2013) [under grant agreement No. 305444 (RD-Connect) to Ł.R.]. The infrastructure was supported by ‘PL-LAB2020’ project, founded by the National Centre for Research and Development, Poland [contract POIG.02.03.01-00-104/13-00].
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}