





Abstract
Genome-wide measurements of paired miRNA and gene expression data have enabled the prediction of competing endogenous RNAs (ceRNAs). It has been shown that the sponge effect mediated by protein-coding as well as non-coding ceRNAs can play an important regulatory role in the cell in health and disease. Therefore, many computational methods for the computational identification of ceRNAs have been suggested. In particular, methods based on Conditional Mutual Information (CMI) have shown promising results. However, the currently available implementation is slow and cannot be used to perform computations on a large scale.
Here, we present JAMI, a Java tool that uses a non-parametric estimator for CMI values from gene and miRNA expression data. We show that JAMI speeds up the computation of ceRNA networks by a factor of ∼70 compared to currently available implementations. Further, JAMI supports multi-threading to make use of common multi-core architectures for further performance gain.
Java 8.
JAMI is available as open-source software from https://github.com/SchulzLab/JAMI.
Supplementary data are available at Bioinformatics online.
1 Introduction
MicroRNAs (miRNAs) are ∼23 nt long RNAs that play an important role in the regulation of transcript abundance in mammalian cells. They are estimated to regulate at least half of the genes in the human genome (Friedman et al., 2009) and thus affect important biological processes and show deregulation in many diseases (Jiang et al., 2009). Several miRNAs often regulate the same transcript in a combinatorial fashion and many transcripts are regulated by the same miRNAs, leading to complex genome-wide networks of co-regulation (Tsang et al., 2010). In these competing endogenous RNA (ceRNA) networks, ceRNA genes that carry binding sites for the same miRNA(s) compete over the limited pool of available miRNA molecules (Arvey et al., 2010; Salmena et al., 2011; Tay et al., 2014). Several examples of ceRNA crosstalk have already been verified, including many genes involved in cancer such as PTEN (Poliseno et al., 2010). This evidence has sparked interest in developing systematic methods for inferring ceRNA interactions from gene and miRNA expression data, reviewed in (Le et al., 2017).
With the emergence of large-scale studies providing gene and miRNA expression data for hundreds of samples, it has become possible to infer ceRNA interactions computationally and several approaches have been suggested to achieve this. Sumazin et al. proposed the use of conditional mutual information in their method HERMES (Sumazin et al., 2011), which was later implemented as part of the CUPID software package (CUPID step III) (Chiu et al., 2015). While this method was applied successfully for inferring ceRNA networks for approximately 450 000 gene pairs (Chiu et al., 2017), the current implementation is very slow and poses a bottleneck for the construction of large-scale networks.
This issue has motivated other researchers to design alternative approaches that are faster. For example methods based on linear correlation (Liu et al., 2017; Paci et al., 2014; Wang et al., 2015). However, in contrast to CUPID, the linearity assumption limits the accuracy of these methods (Le et al., 2017). We thus sought to speed up the computations of CMI values as the only known non-linear alternative for facilitating the efficient construction of large-scale ceRNA networks.
2 Results and discussion
Here, we present JAMI, a novel implementation of the CMI computation step of CUPID (Chiu et al., 2015). Like CUPID, JAMI uses adaptive partitioning for estimating CMI values (Darbellay and Vajda, 1999). This non-parametric estimator is consistent and makes no assumption on the distribution of the data and can thus be used with expression data from any technology. JAMI uses efficient data structures in Java to implement the three-dimensional data partitioning for the computation of CMI values. In contrast to CUPID, JAMI was carefully designed to support multi-threading (Supplementary Fig. S1). In Figure 1, we show that JAMI achieves a substantially better single-threaded runtime compared to CUPID implemented in either Matlab or Java. For the latter comparison, we carefully re-implemented the original CUPID method in Java.
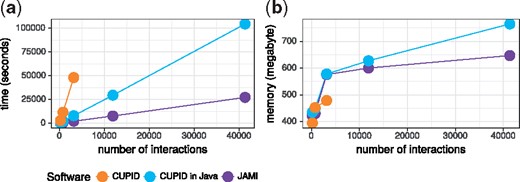
Performance comparison between JAMI, CUPID (Matlab) and CUPID (Java). (a) Process user time in seconds. (b) Peak memory usage
Both JAMI and CUPID rank expression values before the CMI computation. In CUPID, all expression values of 0 are assigned different ranks. This introduces bias and results in positive CMI values even if genes are not expressed in any sample. To avoid this, we extended JAMI to be zero expression aware, and demonstrate that this has considerable effect on the results (Supplementary Figs S2–S5).
Preparing the input for CUPID is tedious and requires separate expression and miRNA interaction files as input for every gene pair of interest. In contrast, JAMI accepts two expression matrices as input, one for gene and one for miRNA expression, and filters these automatically for the data needed. In addition, JAMI offers great flexibility with regards to defining the triplets of interest, making it much more convenient to use JAMI in settings where several genes are of interest. JAMI output files can be directly imported in network analysis tools such as Cytoscape (Shannon et al., 2003). Moreover, JAMI does not require an expensive Matlab® license like CUPID, making it available to a broader audience. To make sure that JAMI can also be used conveniently in a scripting language, we implemented the RJAMI wrapper package for R (http://github.com/SchulzLab/RJAMI).
We illustrate the potential of JAMI by constructing a ceRNA interaction network from the TCGA breast cancer data set (TCGA, 2012) for known ceRNAs (Tay et al., 2014) (Supplementary Fig. S6, see user manual for a step by step guide). The resulting network appears to be much denser than what is reported in the literature, emphasizing the importance of robust tools for ceRNA network inference from widely available expression data.
An open question in the field is whether linear or non-linear methods are better suited for ceRNA network inference (Le et al., 2017). Answering this question was thus far impeded by the lack of a fast tool for computing CMI values. JAMI overcomes this research barrier and facilitates comparisons with correlation-based method such as sensitivity correlation (Paci et al., 2014) (Supplementary Fig. S7).
In conclusion, JAMI is a fast, freely available and well-documented (http://jami.readthedocs.io/) tool primarily targeted at the inference of ceRNA networks. However, its implementation is general and may be used to study other modulators of gene–gene interactions, e.g. transcription factors (Flores et al., 2013).
Funding
This work was supported by the Cluster of Excellence on Multimodal Computing and Interaction (EXC284 to M.S. and J.V.) of the German National Science Foundation (D.F.G.).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Andrea Hornakova and Markus List authors should be regarded as Joint First Authors.


{kind=link}