





Abstract
The fixation index FST can be used to identify non-neutrally evolving loci from genome-scale SNP data across two or more populations. Recent years have seen the development of sophisticated approaches to estimate FST based on Markov-Chain Monte-Carlo simulations. Here, we present a vectorized R implementation of an extension of the widely used BayeScan software for codominant markers, adding the option to group individual SNPs into pre-defined blocks. A typical application of this new approach is the identification of genomic regions, genes, or gene sets containing SNPs that evolved under directional selection.
The R implementation of our method, which builds on the powerful population genetics and genomics software PopGenome, is available freely from CRAN.
Supplementary data are available at Bioinformatics online.
1 Introduction
Genomic analyses that calculate FST values frequently aim to separate neutral contributions to FST caused by population histories from locus-specific contributions due to non-neutral evolution. However, different populations have different histories and are influenced by different factors that affect mutations, genetic drift, and migration (Kronholm et al., 2010). As a consequence, the neutral distribution of FST values strongly depends on the organisms and populations considered. The separation between non-neutral and neutral (population history-based) FST contributions can be made explicit in Bayesian approaches that employ Markov-Chain Monte-Carlo (MCMC) methods to estimate FST (Beaumont and Balding, 2004; Foll and Gaggiotti, 2008; Villemereuil and Gaggiotti, 2015). The BlockFeST method introduced in this paper is based on the work of Beaumont and Balding (2004), which establishes an FST based hierarchical Bayesian model to detect loci that evolve non-neutrally. This Bayesian approach uses a logistic regression model to distinguish between locus-specific effects (selection) and population-specific effects (reflecting the common history of all loci). Foll and Gaggiotti, 2008, extended this work through a reversible jump MCMC method (Green, 1995), which calculates an explicit posterior probability for non-neutral evolution at each locus (implemented in BayeScan, http://cmpg.unibe.ch/software/BayeScan). Whereas these previous algorithms allowed the analysis of either haplotypes or of individual single nucleotide polymorphisms (SNPs), we propose to group neighboring SNPs that likely experienced the same evolutionary pressures into blocks, e.g. by gene, by gene set or by sliding window. This joint analysis of co-evolving SNPs increases the statistical power of the approach, allowing us to more reliably detect directional population-specific selection.
2 Materials and methods
Here, measures the differentiation between subpopulation j and the ancestral population at locus i within region I. αI is a parameter specific to region I (capturing region-specific deviations from neutral evolution), while βj is a parameter specific to population j (capturing locus-independent effects, i.e. consequences of population history). We consider the likelihood contribution of each SNP separately, but assume that the locus-specific effect αI is identical for all SNPs located in the same DNA block I. This means that also in the jump model, all SNPs in one block would be classified as evolving either neutrally () or non-neutrally (). Note that in this modified model, the ancestral population is still modelled as a Dirichlet distribution that considers the allele frequencies of each SNP independently. Recent studies have shown that the reversible jump model tends to produce false positives, particularly for balancing selection (Lotterhos and Whitlock, 2014; Luu et al., 2017). Thus, we restrict the model to the detection of positive (directional) selection. Moreover, in previous simulations (Pfeifer, 2014), we found that the FST values obtained after the initial pilot runs used by BayeScan to tune the reversible-jump model are almost as informative as the corresponding posterior values. Pilot runs are initial MCMC iterations (without the reversible jump model) to estimate the distribution of the model parameters α and β. Thus, we propose a simple sampling scheme to verify significant outlier loci subject to local adaptation based on the distribution of the αI values observed after the MCMC iterations (approximated through a region-specific normal distribution ) without using a time consuming reversible jump model for testing the null hypotheses: (i) For each region I, sample two values , resulting in two distributions of sampled values Dx and Dy across regions. (ii) For each region I, increment its counter cI if yI is above the q-quantile for Dx. (iii) Repeat (i–ii) Niter = 1000 times.
3 Simulation and results
To validate our algorithm and to compare it to alternative approaches, we performed extensive simulations using the MSMS program (Ewing and Hermisson, 2010), which allows coalescent simulations for a structured population under selection. We assume a population with an effective population size of Ne=10 000 that split into two subpopulations 4000 generations ago. After the splitting event, there is no migration between the two populations (island model). To evaluate the performance of the methods to detect selection in cases of variable pairwise population differentiation, we also study a three-population model including an ancestral population (divergence mode) (see Supplementary Material for details and the MSMS calls). We assessed the performance of BlockFeST and compared it to those of BayeScan (Foll and Gaggiotti, 2008) applied to individual SNPs, FST (Hudson, 1992), and the recently published PCA method implemented in the R-package pcadapt (Luu et al., 2017) (see Supplementary Material). For pcadapt and BayeScan, we used the P-values and calculated the sum of logs for each region to compare it to FST and BlockFeST.
We explored a wide range of simulation scenarios, varying recombination rates, selection strengths and coalescent times. Specifically, we studied the robustness of the approaches regarding the effect of neutrality eroding the signal of selection (Supplementary Figs S3–S4 and S8–S9). While FST and pcadapt are the computationally fastest algorithms (Supplementary Table S1), we found that BlockFeST is most powerful and accurate in almost all cases, especially when the signal of local adaptation is not yet eroded by recombination, i.e. in cases of recent positive selection (Fig. 1 and Supplementary Figs S1–S9). For the divergence model, pcadapt and BlockFeST perform comparably well. While the measure of precision shows slightly better results for pcadapt in the divergence model simulations, we obtain higher AUC values for BlockFeST also here, indicating a better balance between sensitivity and specificity (Fig. 1B and Supplementary Figs S4–S9). BayeScan produces elevated P-values, falsely pointing to balancing selection even when no such signal is present, confirming the results in Lotterhos and Whitlock, 2014. This behaviour highlights the tendency of the jump model to produce false positives especially in cases of hierarchical population structures. Despite its simplicity, FST performs well in comparison to the computationally more involved alternatives, with good detection of intermediate signals of positive selection. Overall, we conclude that BlockFeST, which groups SNPs into blocks and calculates empirical P-values, provides faster and more reliable detection of region-specific directional selection compared to the standard application of BayeScan, and performs favourably also compared to alternative approaches.
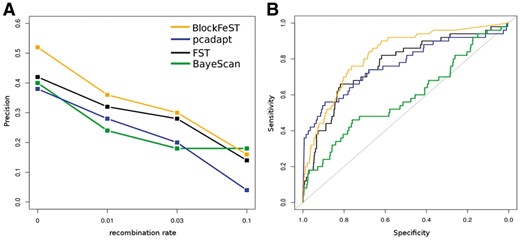
(A) Island model. BlockFeST, pcadapt, FST, and BayeScan are compared in terms of precision, defined as the fraction of true positives recovered in the top 5% of predictions across varying recombination rates. (B) Divergence model. ROC curves for a three-population model, where population 1 and 2 split 4000 generations ago, when directional selection set in, after having split from the ancestral population 8000 ago; here, the recombination rate is set to zero. For a summary of additional simulation results, see Supplementary Figures S1–S9
Acknowledgements
We thank Pablo E. Verde, Gregory Ewing, Stefan Habenschuss, Laura Rose and Juliette de Meaux for helpful discussions.
Funding
M.J.L. acknowledges financial support from the German Research Foundation (DFG grants EXC 1028 and CRC 680).
Conflict of Interest: none declared.
References


{kind=link}