





Abstract
Cancer genomes are altered by various mutational processes and, like palimpsests, bear the signatures of these different processes. The Palimpsest R package provides a complete workflow for the characterization and visualization of mutational signatures and their evolution along tumor development. The package covers a wide range of functions for extracting both base substitution and structural variant signatures, inferring the clonality of each alteration and analyzing the evolution of mutational processes between early clonal and late subclonal events. Palimpsest also estimates the probability of each mutation being due to each process to predict the mechanisms at the origin of driver events. Palimpsest is an easy-to-use toolset for reconstructing the natural history of a tumor using whole exome or whole genome sequencing data.
Palimpsest is freely available at www.github.com/FunGEST/Palimpsest.
Supplementary data are available at Bioinformatics online.
1 Introduction
Mutational signature analysis is a powerful approach to understand the origin of somatic mutations in cancer (Alexandrov et al., 2013). This approach can be extended to any kind of genomic alteration including structural variants (Helleday et al., 2014). Mutational processes may evolve over time, giving different mutational signatures in early clonal and late subclonal mutations (Nik-Zainal et al., 2012; Rubanova et al., 2018). Next-generation sequencing data also allow to reconstruct the timing of copy-number alterations (CNAs) by distinguishing clonal/subclonal events and using the proportion of duplicated mutations to time chromosome duplications (Greenman et al., 2012). Finally, different mutational processes have different propensities to target specific loci depending on the local chromatin state (Polak et al., 2015; Sabarinathan et al., 2016) and sequence context (Letouzé et al., 2017). Understanding how mutational processes evolve during tumorigenesis and alter specific driver genes is critical to better understand the natural history of cancers and eventually predict and influence their clinical behavior.
To the best of our knowledge, no comprehensive workflow integrates mutational signature and clonality analyses to reconstruct the natural history of a tumor. In addition, no automated tools for extracting structural variant signatures, timing chromosome duplications and estimating the process at the origin of each mutation are currently available. The Palimpsest R package provides an extensive toolset to explore and visualize the diverse processes generating somatic alterations in a tumor, their evolution along tumorigenesis and their interaction with driver genes.
2 Features of palimpsest
Palimpsest takes as input somatic mutations and structural variants (optional), copy-number data and a minimal sample annotation file indicating gender and tumor purity. Figure 1A and Supplementary Figure S1 illustrate a typical Palimpsest analysis, as described step by step below.
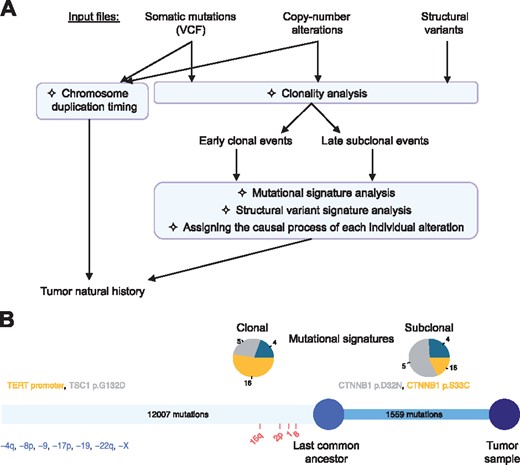
(A) Workflow of a typical analysis with Palimpsest. (B) Oncogenic timeline of a tumor representing the number of clonal/subclonal mutations, the contribution of mutational processes to each step and the timing of driver mutations (colored according to their most likely causal mutational process) and CNAs
Mutational signature analysis. Palimpsest allows both de novo extraction of novel mutational signatures or quantification of previously described signatures using non-negative matrix factorization (NMF), as implemented in the NMF R package (Gaujoux and Seoighe, 2010). Utilities are provided to visualize signatures, transcriptional strand biases and inter-mutation distance, and to compare signatures using cosine similarity.
Structural variant signature analysis. Palimpsest implements an adaptation of the mutational signature analysis framework for structural variants (SVs). SVs are first classified into 38 categories according to the type (deletion, tandem duplication, inversion and interchromosomal translocation) and size of rearrangements (Nik-Zainal et al., 2016). NMF then allows extracting new or known structural variant signatures and their contribution to each tumor genome.
Predicting the process at the origin of each individual mutation. Once the mutation catalogue of a tumor has been deconvoluted as the addition of several mutational processes, Palimpsest estimates the probability of each individual mutation being due to each process using simple Bayesian statistics (Letouzé et al., 2017). This key feature, available for both point mutations and structural variants, allows predicting the processes most likely at the origin of driver events in each tumor.
Clonality analysis and evolution of mutational signatures. Palimpsest integrates variant allele fraction, tumor purity and local copy number to estimate the proportion of tumor cells harboring each mutation and classify them as clonal or subclonal. Then, the package extracts and compares mutational signatures operative in clonal and subclonal mutations to explore the evolution of mutational processes along tumorigenesis.
Timing CNAs. Several tools have been described to differentiate clonal from subclonal CNAs, which can be directly specified in the CNA input file. In addition, Palimpsest will estimate the molecular timing of duplications using the proportion of duplicated/non-duplicated somatic mutations.
Oncogenic timelines. Finally, Palimpsest integrates the results of clonality and mutational signature analyses to generate a comprehensive representation of the natural history of the tumor (Fig. 1B).
3 Conclusions
Unraveling what mutational processes generate driver mutations and fuel the initiation and progression of tumor clones is crucial to better understand the natural history of cancers and optimize personalized patient care. Palimpsest integrates a broad range of functions within a single convenient workflow to provide a complete picture of tumorigenesis. The minimum basic input data make the package easy-to-use in any cancer genomic study downstream classical variant calling and copy number analyses.
Acknowledgements
We thank the CIT program from the French Ligue Nationale Contre le Cancer for providing useful R codes (cit.x functions).
Funding
This work was supported by INSERM (“Cancer et environnement” & HTE program), cancéropôle Ile-de-France (exhauTrans project), Région Ile-de-France, BPI France (ICE project), CARPEM and ICGC. The group is supported by the Ligue Nationale contre le Cancer (Equipe Labellisée), Labex OncoImmunology Investissement d’Avenir and Fondation Mérieux.
Conflict of Interest: none declared.
References


{kind=link}