





Abstract
Predicted deleteriousness of coding variants is a frequently used criterion to filter out variants detected in next-generation sequencing projects and to select candidates impacting on the risk of human diseases. Most available dedicated tools implement a base-to-base annotation approach that could be biased in presence of several variants in the same genetic codon. We here proposed the MACARON program that, from a standard VCF file, identifies, re-annotates and predicts the amino acid change resulting from multiple single nucleotide variants (SNVs) within the same genetic codon. Applied to the whole exome dataset of 573 individuals, MACARON identifies 114 situations where multiple SNVs within a genetic codon induce an amino acid change that is different from those predicted by standard single SNV annotation tool. Such events are not uncommon and deserve to be studied in sequencing projects with inconclusive findings.
MACARON is written in python with codes available on the GENMED website (www.genmed.fr).
Supplementary data are available at Bioinformatics online.
1 Introduction
Variant annotation is a crucial step in whole genome/exome sequencing analyses aimed at identifying putative causal variants, especially in a clinical context (Ding et al., 2014). For example, for a rare inherited disease, one often starts to filter out detected variants according to the anticipated mode of inheritance, the type of variations (e.g. synonymous, non-synonymous, stop gain/loss, splice, etc.), allele frequencies and their predicted deleteriousness. There is a plethora of annotation tools (Cingolani et al., 2012; McLaren et al., 2016; Yang and Wang, 2015) but most of them implement a base-to-base approach to annotate single-nucleotide variants (SNVs). However, the presence of several SNVs at the same locus, in particular within the same genetic codon, may bias annotations. For example, two synonymous SNVs in the same codon can generate a non-synonymous variation that would be missed by standard annotation tools. To our knowledge, there is only one program, MAC (Wei et al., 2015), that accommodates multiple SNVs simultaneously. However, it is restricted to adjacent SNVs and cannot then properly address the situation when two SNVs affect the first and the third base of a genetic codon. In addition, it does not use the information on genetic code triplet structure. As a consequence, it considers the same way two SNVs affecting the adjacent bases of a genetic codon, and two SNVs affecting the last base of a codon and the first base of the next codon. To fill these gaps, we propose a simple python-based algorithm, MACARON (for Multi-bAse Codon-Associated variant Re-annotatiON) to identify and to more accurately annotate multiple SNVs occurring within the same genetic codon (Fig. 1). We illustrate MACARON's relevance by an application to whole exome sequencing data of 573 subjects.
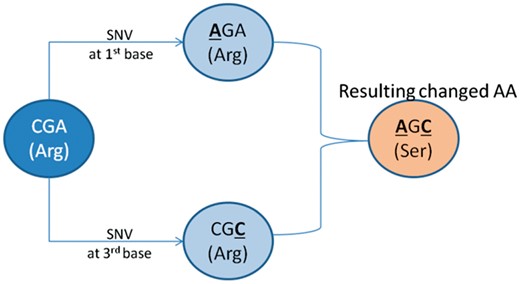
Illustration of the impact of the presence of two single nucleotide variations within the same genetic codon on the resulting amino acid change
2 Implementation and application
2.1 Workflow
The overall algorithmic steps of MACARON are given below and illustrated as Supplementary Figure S1. The algorithm of MACARON is written in python language and can run on any LINUX/UNIX-like environment. Two pre-installed software, GATK (McKenna et al., 2010) and SnpEff (Cingolani et al., 2012) should be available for a complete run of MACARON. Briefly, MACARON starts with a VCF file as an input with no restriction on file format specifications. After identifying a list of candidate SNVs that occur within the same genetic codon along with their corrected amino acid changes, a second step consists in reading through the original BAM files to extract reads information and to confirm the presence of multiple SNVs on the same reads.
First, starting with a VCF file, MACARON utilizes GATK's VariationFiltration walker (Van der Auwera et al., 2013) with parameters of –clusterSize 2 and –clusterWindowSize 3 followed by the SelectVariants tool to identify adjacent SNVs and SNVs that are 2 bps apart. Then, coding SNVs are selected based on the SnpEff functional annotation classes: SILENT, MISSENSE and NONSENSE (temp_file1). At the third step, SNVs that cluster within the same genetic codon are kept and new amino acid (AA) changes are written in temp_file2 and temp_file3. Next, clustered SNVs whose resulting AA changes are different from the original ones are stored in temp_file4. In case of a multi-sample VCF file, a scan is then performed on temp_file4 to identify clustered SNVs that are present in at least one individual. Results are stored in a final output text file containing all those SNVs identified within the same genetic codon and for which the allelic status is heterozygous or homozygous compared to the reference. At the final step, in order to confirm that identified clustered SNVs are harbored on the same reads, we used an in-house BASH-shell script (available with MACARON code) to read through the original BAM files that have been used for VCF file generation and to report the number of reads that harbor all variant alleles at the identified clustered SNVs. This script needs a subset of BAM files covering 50 bps over each clustered SNVs.
2.2 Results
MACARON was applied to the whole exome sequencing data of 573 healthy individuals as part of the FREX initiative in which 625 984 exonic SNVs were identified (Genin et al., 2017). MACARON identified 114 multi-base affected codons in 194 participants. All identified affected codons were impacted by two SNVs (these were referred to as paired codon SNVs, pcSNVs) and no codon was identified that was simultaneously affected at all its 3 bases. From the identified pcSNVs, 83 were affecting codon positions 1 and 2, 23 codons were affected at positions 2 and 3 and the remaining 8 were affected at positions 1 and 3. Detailed distribution of the identified pcSNVs according to different criteria including allele frequencies, amino acid changes and predicted deleteriousness is given in Supplementary Table S1. Several observations could be made. For example, of these pcSNVs, 30 involved two rare [i.e. never reported or reported with minor allele frequency <0.01 in the gnomeAD database (Lek et al., 2016)] SNVs, 15 involved one rare and one common SNV and 69 based on two common SNVs. These types of pcSNVs were referred to as ‘double-rare’, ‘single-rare’ and ‘double-common’ pcSNVs, respectively. The number of private (i.e. present in only one individual) pcSNVs were 16 (53%), 11 (∼73%) and 3 (∼4%) ∼ among ‘double-rare’, ‘single-rare’ and ‘double-common’ pcSNVs, respectively. No pcSNV was generated from two synonymous SNVs but 26 were defined from one synonymous and one non-synonymous SNV. For 114 pcSNVs, the resulting amino acid change was different from the two original SNVs. Using the popular functional effect prediction tool SIFT (Ng and Henikoff, 2003), we observed that nine pcSNVs were predicted to be ‘damaging’ while the two original SNVs were predicted to be ‘tolerated’. Conversely, two pcSNVs were predicted to be ‘tolerated’ or ‘neutral’ while the two original SNVs were predicted to be ‘damaging’. For this application, MACARON took ∼1 h on an Intel(R) Xeon(R) CPU E5-2640 v3 @ 2.60 GHz processor ×32 cores machine equipped with 64 GB of RAM on UBUNTU 16.04 LTS operating system to screen, re-annotate pcSNVs and validate them from BAM files.
3 Conclusion
MACARON is a new annotation tool for characterizing multiple SNVs within a same codon detected in WGS/WES studies. Its application to real data suggests that the frequency of pcSNVs is underappreciated and that inaccurate annotation of such genetic variations could contribute to explain inconclusive findings in DNA sequencing analyses.
Acknowledgements
Members of the GENMED and FREX consortia are listed in supplements.
Funding
This work was supported by the GENMED Laboratory of Excellence on Medical Genomics [ANR-10-LABX-0013 to WK, GV-S, FT] and the France Genomique National Infrastructure [ANR- 10-INBS-0009 to FREX consortium].
Conflict of Interest: none declared.
References


{kind=link}