





Abstract
The traditional view of cancer evolution states that a cancer genome accumulates a sequential ordering of mutations over a long period of time. However, in recent years it has been suggested that a cancer genome may instead undergo a one-time catastrophic event, such as chromothripsis, where a large number of mutations instead occur simultaneously. A number of potential signatures of chromothripsis have been proposed. In this work, we provide a rigorous formulation and analysis of the ‘ability to walk the derivative chromosome’ signature originally proposed by Korbel and Campbell. In particular, we show that this signature, as originally envisioned, may not always be present in a chromothripsis genome and we provide a precise quantification of under what circumstances it would be present. We also propose a variation on this signature, the H/T alternating fraction, which allows us to overcome some of the limitations of the original signature.
We apply our measure to both simulated data and a previously analyzed real cancer dataset and find that the H/T alternating fraction may provide useful signal for distinguishing genomes having acquired mutations simultaneously from those acquired in a sequential fashion.
An implementation of the H/T alternating fraction is available at https://bitbucket.org/oesperlab/ht-altfrac.
Supplementary data are available at Bioinformatics online.
1 Introduction
The development of cancer is driven by the accumulation of somatic mutations within a set of cells. These mutations can vary from single nucleotide variants to large-scale structural variations (SVs)—including deletions, duplications, inversions and translocations—that rearrange entire segments of DNA. Although the traditional view of cancer evolution states that a cancer genome accumulates a sequential ordering of these mutations over a long period of time (Nowell, 1976), Stephens et al. (2011) proposed an alternative model where instead a collection of rearrangements occur simultaneously. Specifically, they characterized chromothripsis as an event where a portion of a chromosome (or several chromosomes) shatter and a subset of the fragments are randomly stitched together. As a result, the participating chromosomes appear highly rearranged—containing a number of closely located and overlapping SVs.
Although the exact underlying mechanisms that drive chromosome shattering in cancer remain unknown, a number of studies have reported the presence of chromothripsis in numerous cancer types including bone, colorectal, and prostate cancers (Fraser et al., 2017; Kloosterman et al., 2011b; Stephens et al., 2011). Often times samples labeled as chromothripsis are associated with a poor outcome for the patient (Kloosterman et al., 2014; Molenaar et al., 2012). Chromothripsis has also been reported in germline samples—usually in conjunction with other complex diseases (Collins et al., 2017; Kloosterman et al., 2011a). Furthermore, other related phenomenons have been suggested that also indicate the accumulation of multiple rearrangements at once. For example, chromoplexy (Baca et al., 2013) is similar to chromothripsis but generally tends to have few aberrations that affect more chromosomes. On the other hand, Liu et al. (2011) suggested using the term chromoanasynthesis instead of chromothripsis in order to better reflect potential mechanisms underlying the phenomenon, but some treat this a separate category of simultaneous events (Collins et al., 2017).
A fundamental question underlying these continuing reports of chromothripsis, and other simultaneous events, is: How does one distinguish a sequential accumulation of mutations from a simultaneous acquisition? The original chromothripsis paper (Stephens et al., 2011) first noted that the genomes they believed to have undergone chromothripsis exhibited some striking patterns: (i) clustering of break points along affected portions of the genome; and (ii) oscillating copy number states. The later observation is a result of genomic segments in the shattered region not being included in the derivative chromosome, and therefore appearing as deleted. The initial discovery (Stephens et al., 2011) and later reports of chromothripsis (Malhotra et al., 2013; Rausch et al., 2012) rely on Monte Carlo simulations to argue that sequential rearrangements were unlikely to produce these patterns.
Subsequently, Korbel and Campbell (2013) built upon these observations and proposed a list of six criteria for chromothripsis: clustering of breakpoints, oscillating copy number states, interspersed loss and retention of heterozygosity, breakpoints affecting one haplotype, randomness amongst fragment join types and ability to walk the derivative chromosome. Although approaches designed to detect complex rearrangements in cancer such as CouGaR (Dzamba et al., 2017), PREGO (Oesper et al., 2012) or McPherson et al. (2012) might be useful for detecting and analyzing chromothripsis, methods designed specifically for this task, including many that rely on a subset of these proposed signatures, may be more appropriate (Baca et al., 2013; Cai et al., 2014; Govind et al., 2014; Weinreb et al., 2014). For instance, ShatterProof (Govind et al., 2014) combines scores across several criteria to identify regions of a genome that are likely to have undergone a chromothripsis event. Cai et al. (2014) identify what they call ‘chromothripsis-like patterns’ or CTLP by clustering copy number status changes (effectively merging two of the signatures). So, while some of the signatures proposed by Korbel and Campbell (in particular those also reported in the original chromothripsis publication) appear to be used frequently in practice, further analysis of the other signatures may provide additional insight.
In this article, we perform an analysis of one of the signatures proposed by Korbel and Campbell, the ability to walk the derivative chromosome. In particular, we show that this signature, as originally envisioned, may not always be present in a chromothripsis genome and we provide a precise quantification of under what circumstances it would be present. We also propose a variation on this signature, the H/T alternating fraction, which overcomes some of the limitations of the original signature. We apply our measure to both simulated data and a previously analyzed real cancer dataset and find that the H/T alternating fraction may be useful for distinguishing genomes having acquired mutations simultaneously from those acquired in a sequential fashion.
2 Materials and methods
2.1 Chromothripsis strings
Consider a unichromosomal reference genome G where we label consecutive intervals, or genomic segments along G using the characters . Let be the set of these characters. An interval that appears in the reverse orientation in a genome derived from G will be denoted using its inverse-g. We define to be the set of all characters in S and their inverses. A linear cancer genome C derived from G via genomic rearrangements is a sequence of characters from . Therefore, , the Kleene closure of (representing the set of all rearrangement strings that can be derived from G). A chromothripsis event rearranges genomic segments, allowing for deletion of some segments, but no duplication of segments. Therefore, only a subset of can be the result of a chromothripsis event. We now define which rearrangement strings in may result from a chromothripsis event.
Definition 2.1. We define a linear string to be a chromothripsis string if C is a signed permutation of 2 or more characters from S.
2.2 Extremities, adjacencies and extremity permutations
We also define , the terminal set of C, as the set of extremities from E appearing in some adjacency in but where the corresponding obverse extremity for the interval does not appear in any adjacency in . Note that an extremity in the terminal set indicates that the associated character (or interval) must appear at the end of the string C, and in terms of genomes may be interpreted as the telomere. We define the extremity permutation to be the unique set of extremities appearing in some element of after sorting them according to their position in G. An extremity permutation is H/T alternating if its sequence of extremities alternate between being members of the sets H and T. In such a circumstance we may also refer to C as being H/T alternating. Figure 1 shows three examples of strings C along with their corresponding adjacency sets , terminal sets and extremity permutations .
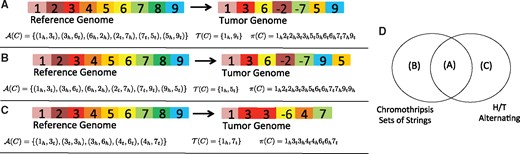
(A–C) Three different tumor genomes obtained as a rearrangement of blocks from the reference genome along with their corresponding adjacency sets, terminal sets and extremity permutations. (D) Shows the relationship of the previous three examples in terms of a string C being a chromothripsis string or having an extremity permutation that is H/T alternating
2.3 H/T alternating chromothripsis strings
We now explore the relationship between our model and the last of the six signatures of chromothripsis suggested by Korbel and Campbell Korbel and Campbell (2013)—‘the ability to walk the derivative chromosome’. They further describe this signature as defined by an alternating head/tail pattern observed when measured tumor adjacencies are sorted according to their position in the reference genome. In our model, this corresponds to being H/T alternating. With our model in place, we can now assess how representative this signature really is of an arbitrary chromothripsis string.
In Theorem 1 we quantify two necessary and sufficient conditions that determine when a chromothripsis string C will have an extremity permutation that is H/T alternating. In particular, the first condition corresponds to when a chromothripsis event occurs somewhere in the middle of a chromosome, leaving the telomeres in place (Fig. 1A), while the second condition corresponds to a degenerate case where both ends of the derivative chromosome originate in a particular configuration from the interior of the chromosome.
Suppose that C is a chromothripsis string for G. is H/T alternating if and only if the terminal set is one of the following:
1. where , and .
2. There exists some k such that where , and .
Proof. Let C be a chromothripsis string for G.
() Assume that is H/T alternating. We will proceed by contradiction. Assume that none of the above conditions about are true. In particular, we can also assume that there exists some such that but . Therefore, there must exist some such that and . However, if , this implies that does not alternate. Similarly, if , this implies that does not alternate—a contradiction. Hence, one of the above conditions about must be true.
() We will consider each possible telomere set separately and show that for each it is true that is alternating.
Assume that where , and . We will proceed by contradiction. Assume that is not alternating. This implies (without loss of generality) that there exists some and such that (that is ). This implies that and therefore . And since , it must be the case that , therefore contradicting our assumption that . The argument for is similar. Hence, must be H/T alternating.
Assume there exists some k such that where , and . We will proceed by contradiction. Assume that is not alternating. This implies (without loss of generality) that there exists some and such that (that is ). This implies that and therefore . There are only two possible values of such that . The first possibility is that . If , then , a contradiction with our assumption that . The second possibility is that . If , then , a contradiction to our assumption that . The argument for is similar. Hence, must be H/T alternating.
It is important to note that the cases detailed in Theorem 1 do not include the case where a chromothripsis event includes one telomere and the final segment joined to form the end of the derivative chromosome is different than the original telomere (Fig. 1B). This is potentially an observation of significance as recent studies have suggested that chromothripsis may arise as the result of telomere crisis (Maciejowski et al., 2015). Thus, even in the absence of noise, a chromothripsis string may not be H/T alternating. Furthermore, a linear string C that is not a chromothripsis string may still have an extremity permutation that is H/T alternating (Fig. 1C).
2.4 Fraction of chromothripsis strings that are H/T alternating
In the previous section we demonstrate that not all chromothripsis strings C have extremity permutations that are H/T alternating and categorize the subset that does exhibit this property. If the vast majority of chromothripsis strings exhibit this signature, then it may still be useful for categorizing chromothripsis. We therefore derive a formula for the exact fraction of chromothripsis strings that are H/T alternating in Theorem 2 (see Supplementary Appendix for proof).
The fraction of chromothripsis strings of length m derived from a reference genome G composed of n intervals with that is H/T alternating is .
Theorem 2 tells us that the fraction of chromothripsis strings that are H/T alternating depends only on the number m of characters in the chromothripsis string and not the original number of intervals n. More importantly, as m increases this fraction decreases quickly (Fig. 2). For example, only 5.6% of chromothripsis strings containing 10 intervals are H/T alternating. Thus, in its current stringent form, the H/T alternating property does not seem well suited as a signature of chromothripsis.
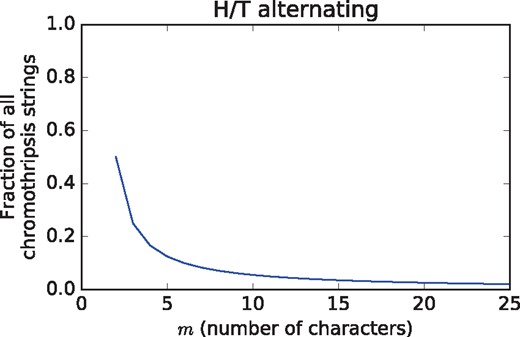
The fraction of chromothripsis strings C that are H/T alternating according to Theorem 2
2.5 An alternative signature: H/T alternating fraction
Treating H/T alternating as a binary feature that a chromothripsis string C either exhibits or it doesn’t, prevents us from capturing any information about how close or far C is from H/T alternating. For example, consider the following two sequences of H/T membership that an extremity permutation of length 8 might exhibit: HTHTHTHH and HHHHTTTT. Neither one is H/T alternating, but the former is much closer to H/T alternating than the later. Thus, it would be useful to have a measure that allows us to capture this type of more nuanced information.
2.6 Distinguishing between simultaneous and one-off events
In the previous sections, we showed that the H/T alternating fraction allows for a more robust version of the signature originally suggested by Korbel and Campbell (2013). Here we consider whether this signature is able to distinguish between one-off events such as chromothripsis and step-wise events.
First we note that a cancer genome C derived from simple, non-overlapping rearrangements may in fact result in a rearrangement string that is a chromothripsis string. For example, consider a reference genome and a cancer genome having undergone an inversion of interval 2. In this instance . In the most extreme case, a non-rearranged cancer genome would also be a chromothripsis string and have . So such genomic rearrangements are technically possible from a chromothripsis event, these configurations seem unlikely to have arisen that way, and we would likely not want to classify them as the result of chromothripsis. One key distinction in these cases is that these simple rearrangements do not include any of the other traditional signatures of chromothripsis, including overlapping rearrangements. Therefore, we will focus on the utility of the H/T alternating fraction when considering overlapping rearrangements where the distinction of chromothripsis versus a step-wise sequence of events is much less obvious.
Next, we consider a different situation. Consider a reference genome and a cancer genome obtained through a tandem duplication of intervals 2, 3 followed by an overlapping deletion of intervals 3, 4. In this instance, C is not a chromothripsis string as it contains a duplication. If we observed the exact sequence of intervals in C we would have and . This would seem problematic for our goal of using H/T alternating fraction to distinguish between one-off and simultaneous events. However, we make the important observation that we don’t actually observe the sequence of intervals in a cancer genome. Instead we observe the set of novel adjacencies (not occurring in the reference genome) between regions of the genome and it is these adjacencies that define the set of genomic intervals used in the derivative chromosome. So, for the above example we would only observe which leads to which has lower .
We note that there are certainly instances where a progressive sequence of overlapping rearrangements will not exhibit such a drop in AF(C). For example, consider a reference genome and a cancer genome obtained through an inversion of intervals 2, 3, followed by a deletion of intervals . In this example we would observe which has . Other progressive sequences of events that include duplications, but also utilize breakpoint reuse, such as a reference genome and cancer genome also have . Thus, the H/T alternating fraction is not a perfect classifier between one-off and simultaneous events. However, it does have the potential to be useful in some situations, as all chromothripsis events would have , compared with only a subset of progressive events.
2.7 Extending to multiple chromosomes
Thus far we have presented this work in terms of a unichromosomal genome G. Extending to the case of a multi-chromosomal genome is straightforward. This is done by considering the set of extremities originating from each chromosome separately. For example, if the extremity permutation for each chromosome is H/T alternating, then entire set is H/T alternating. Similarly, the H/T alternating fraction is computed by considering only adjacent pairs of extremities appearing within the same chromosome. In the ideal situation we would phase the rearrangements first, and consider each haplotype separately. Given the difficulty with phasing rearrangements, this may not be feasible and instead we consider a union of the rearrangements on the two chromosomes. If overlapping rearrangements have occurred to both copies of the chromosome, this can affect the reported AF(C) values, both dropping AF(C) for true chromothripsis events and raising AF(C) for progressive events.
3 Results
We demonstrate the usefulness of the H/T alternating fraction measure on both simulated and real DNA-sequencing data.
3.1 Simulations
We simulate data by shattering a reference genome into n blocks and then create random chromothripsis genomes that have m novel adjacencies between those blocks. We introduce noise into the set of adjacencies by either removing true adjacencies or adding extraneous adjacencies to the set (see Supplementary Appendix for further details).
For different values of m we verified that our observed fraction of chromothripsis genomes that are strictly H/T alternating matched well with our theoretically derived fraction (Theorem 2). These fractions are quite low to begin with (e.g. only 3.5% of 10 000 random genomes with m = 15 adjacencies are H/T alternating) and this fraction becomes even smaller as noise is added (see Supplementary Appendix). Therefore, we restricted our attention to chromothripsis genomes that are originally H/T alternating, and even in that instance we observe that the H/T alternating signature degrades quickly with noise (see Supplementary Appendix). For example, <3% of 10 000 simulations with m = 15 novel adjacencies are H/T alternating when just a single adjacency is either added or removed. We do note that Korbel and Campbell (2013) indicate that the version of this signature they propose is dependent on observing all adjacencies. In contrast, we show that the H/T alternating fraction measure AF(C) is much more robust to noise (Fig. 3). For example, over simulations with m = 35 novel adjacencies in the chromothripsis genome, adding or removing up to four adjacencies produces an alternating fraction AF(C) above 0.9 and 0.87, respectively, in all 10 000 random trials.
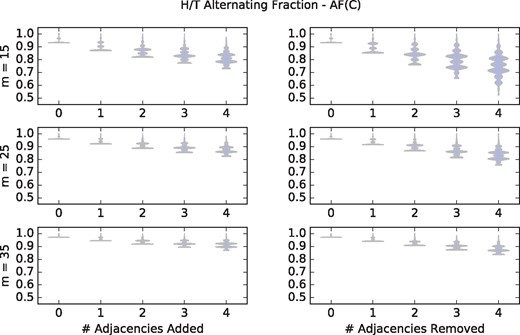
Violin plots over 10 000 randomly generate chromothripsis genomes (with m novel adjacencies) showing that the alternating fraction AF(C) measure remains high even when noise is incorporated by randomly adding and removing adjacencies
The H/T alternating fraction measure is more useful if it can distinguish chromothripsis genomes from genomes that have undergone a sequential accumulation of events. Therefore we simulated genomes that have undergone a sequential accumulation of random events including deletions, tandem duplications and inversions. We created these genomes by randomly adding such events to a reference genome until the number of novel adjacencies was equal to or exceeded a specified value m (see Supplementary Appendix for further details). We compare the observed distribution of H/T alternating fractions for chromothripsis genomes and sequential genomes (Fig. 4) and find that in the case of noise-free data, the H/T alternation fraction or AF(C) value for chromothripsis genomes is much higher than for the sequential genomes. We also created simulated data where where mutations occur as part of an evolutionary branching process instead of a single step-wise accumulation of mutations (see Supplementary Appendix For Further Details) and found the results to be very similar to those presented here (see Supplementary Appendix). It is important to note that the observed difference in H/T alternating fraction between the one-off genomes and the step-wise genomes shown in Figure 4 will diminish as noise is added into the data. In particular, the corresponding panel in Figure 3 with m = 25 shows that noise will lower the H/T alternating fraction for the one-off genomes, but will still remain largely above what we observe here for the step-wise genomes. See the Supplementary Appendix for a direct comparison. Thus, these results indicate that the H/T alternating fraction does indeed capture some quantifiable aspect about the presence of chromothripsis. Although certainly not perfect—some sequentially generated genomes may also have a high H/T alternating fraction—the signature appears applicable in some instances.
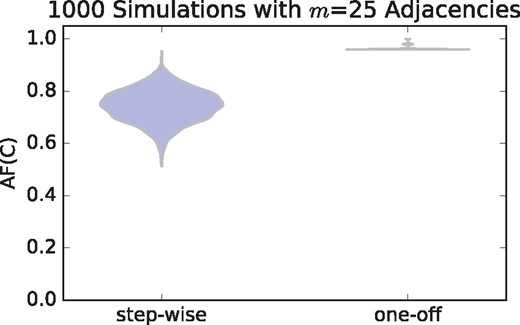
In error-free simulations, we observe that the H/T alternating fraction AF(C) measure is much higher for one-off (chromothripsis) genomes than genomes that have undergone a step-wise sequence of events and have the same number of novel adjacencies
3.2 Real data
We apply our H/T alternating fraction measure to a dataset of 64 genomes representing seven tumor types from the The Cancer Genome Atlas (TCGA) that were previously analyzed for chromothripsis by Malhotra et al. (2013). In particular, they identify 154 sets of observed adjacencies that they classify as either one-off (chromothripsis) events (97 sets of adjacencies) or as step-wise (57 sets of adjacencies). We compute the H/T alternating fraction across each of these sets (Fig. 5A) and find that the mean of the one-off set is 0.689 and the mean of the step-wise set is 0.602 with a difference in means of 0.087. We use a permutation test with 100 000 permutations to assess the statistical significance of the observed difference in means between the two sets, resulting in a permutation distribution with mean of and standard error of 0.032, and find that the H/T alternating frequency of the one-off events is statistically higher than the step-wise events (P = 0.00265). We also compute a bootstrap-t CI over 10 000 replicates and determine that with 95% the average one-off H/T alternating fraction is between 0.029 and 0.15 U larger than the average step-wise H/T alternating fraction. For additional statistical analysis of the differences between these distributions, see the Supplementary Appendix.
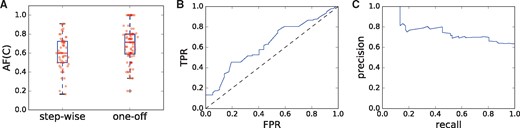
(A) Sets of adjacencies previously classified by Malhotra et al. (2013) as one-off (chromothripsis) have a statistically higher observed H/T alternating fraction AF(C) that those sets classified as step-wise (P = 0.00265 with a permutation test). (B) ROC curve (AUC = 0.63) and (C) precision-recall curve (AUC = 0.76) when events labeled as one-off are considered the positive class and the threshold for classifying as one-off or step-wise based on AF(C) is varied
We also analyze the use of different thresholds t of the H/T alternating fraction, or AF(C), to classify a sample as one-off [AF(C) ≥ t] or step-wise [AF(C) < t]. In particular we see that AF(C) serves as a modest classifier by considering the associated ROC curve (AUC = 0.63) and precision-recall curve (AUC = 0.76) as shown in Figure 5B and C, respectively. Although these results are not strong enough to support the use of the H/T alternating fraction as a classifier in isolation, they do indicate that this signature does provide some signal and may therefore lead to improved results when used in conjunction with other signatures to do classification.
We note that our computations of H/T alternating fraction also allows us to determine which, if any of these samples, exhibit the strict H/T alternating criterion as they would have a value of AF(C) = 1.0. We find that only 13 of the 154 sets of adjacencies have AF(C) = 1.0, all of which were originally classified as one-off. However, we also note that these sets contain relatively few breakpoints, only six or eight in each one, the smallest values contained in any sets in the entire dataset.
Weinreb et al. (2014) later re-analyzed this dataset and identified some sets of adjacencies that may have originally been misclassified. This includes lung squamous cell carcinoma sample LUSC-11 (adjacency set chain-5) which Malhotra et al. originally classified as step-wise but Weinreb et al. suggest that it may indeed be the result of a one-off event like chromothripsis. We find a relatively high H/T alternating fraction value of 0.73 for this sample. Although this is not the highest H/T alternating fraction found for any set of rearrangements originally labeled as step-wise (in fact it is the 12th highest), this set of rearrangements contains 84 breakpoints along a single chromosome, which is many more than the 11 step-wise rearrangement sets with higher H/T alternating fractions that have on average only 14.7 breakpoints. Observing a high H/T alternating fraction with more breakpoints may provide additional confidence that the pattern is not due to chance. We interpret these results as further supporting evidence that this sample may indeed have originally been misclassified.
4 Discussion
Determination of a rigorous signature that may be useful for identification of a one-time event, such as chromothripsis, has proven to be a challenging task (Kinsella et al., 2014). A number of different criteria for inference of chromothripsis have been proposed (Stephens et al., 2011) including a list of six signatures proposed by Korbel and Campbell (2013). Although some of these signatures (e.g clustering of breakpoints, oscillation of copy-number states) have been widely used in practice (Malhotra et al., 2013; Govind et al., 2014; Rausch et al., 2012) others have been less studied. In this work we provide a rigorous formulation and analysis of the ‘ability to walk the derivative chromosome’ signature proposed by Korbel and Campbell (2013), which we refer to as H/T alternating. In particular, we have shown that this signature, as originally envisioned, may not always be present in a chromothripsis genome and we provide a precise quantification of under what circumstances it would be present. We then propose a variation on this signature, the H/T alternating fraction, which allows us to measure to what degree the H/T alternating property, originally defined by Korbel and Campbell (2013), is present throughout the genome. We apply this measure to a previously analyzed dataset and find that sets of rearrangements previously classified as one-off (chromothripsis) have a statistically higher H/T alternating fraction than those classified as step-wise. Thus, indicating that the H/T alternating fraction may be an indicative measure of rearrangements obtained simultaneously as opposed to sequentially.
Although many studies have investigated the occurrence of chromothripsis in different tumor types, the relative prevalence of the phenomenon remains unknown (Rode et al., 2016). Some studies have attempted to estimate the rate of chromothripsis by considering large datasets utilizing existing methods or signatures to identify likely chromothripsis candidates. For example, Cai et al. (2014) identified 918 cancer samples with chromothripsis-like patterns from a dataset of >22 000 cases. This yields a relatively low prevalence rate of only 4.2%. This is similar to the 2–3% prevalence rate estimated in the original Chromothripsis publication (Stephens et al., 2011). However, despite low prevalence across all tumor types, the ability to accurately determine these instances has important potential impact as samples labeled as chromothripsis are often associated with poor patient outcome (Kloosterman et al., 2014; Molenaar et al., 2012). Furthermore, the estimated rate of chromothripsis is much higher in some types of cancer. For example, Stephens et al. (2011) estimated a rate of 25% in bone cancers. Thus, improved methods for identifying genomes that may have undergone a catastrophic event like chromothripsis is an important and necessary step in the search to better understand and treat cancer patients.
There are a number of avenues for further investigation relating to this work. In particular, we have demonstrated the applicability of the H/T alternating fraction on one curated dataset where sets of rearrangements had already been classified as either one-off (chromothripsis) or step-wise. Many other chromothripsis studies do not include sets of rearrangements classified as step-wise in addition to those classified as chromothripsis, making it difficult to assess how well this signature generalizes to other real datasets. We did calculate the H/T alternating fraction for 15 additional genomes classified as chromothripsis by two additional studies by Rausch et al. (2012) and Stephens et al. (2011). But since both of these studies did not include genomes having undergone similar processing and called as step-wise, it is difficult to appropriately assess the meaning of their H/T alternating fractions. To this end, we did add these genomes to the set classified by Malhotra et al. (2013) as one-off and compared them to the set classified as step-wise, and still find a significant difference in the H/T alternating fraction of the two groupings (P = 0.0036 with a permutation test comparing the difference of the means of the two groupings). Further study of this signature applied to new datasets as they become available may yield further insight into its usefulness.
Another important area for further investigation is how this signature may be the most useful in practice for classifying rearrangements as sequential or simultaneous. In this work we have focused mainly on analyzing the ‘ability to walk the derivative chromosome signature’ proposed by Korbel and Campbell (2013) and one variation of this signature in a largely proof-of-concept context. Given that we have shown that the H/T alternating fraction degrades with noise and is dependent on the number of adjacencies involved in the rearrangement, we suspect that this signature may be most useful when used in combination with other proposed signatures of chromothripsis, such as clustering of breakpoints or oscillation of copy number, rather than in isolation. Exploration of how to integrate this signature with other signatures in order to detect chromothripsis events, perhaps in the context of new or existing tools such as ShatterProof (Govind et al., 2014), is left as future work.
Acknowledgements
We thank Vianne Gao for the useful conversations related to this project.
Funding
L.O. is supported by NSF CRII award [IIS-1657380], S.D. is supported by CAPES Project ID 18316-12-3, CNPq and FAPERJ and B.J.R. is supported by NSF CAREER Award [CCF-1053753] and US National Institutes of Health (NIH) grants [R01HG007069 and U24CA211000].
Conflict of Interest: B.J.R. is a co-founder and consultant at Medley Genomics.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}