





Abstract
Medical Subject Headings (MeSH) is the NLM controlled vocabulary used to manually index articles for MEDLINE/PubMed. MeSH provides unique and comprehensive annotations for life science. The meshes package implements measurement of the semantic similarity of MeSH terms and gene products to help using MeSH vocabulary in knowledge mining. Enrichment analysis to extract the biological meanings from gene list, expression profile and genomic regions is also provided using MeSH annotation. Meshes supports more than 70 species and provides high quality visualization methods to help interpreting analysis results.
meshes is released under Artistic-2.0 License. The source code and documents are freely available through Bioconductor (https://www.bioconductor.org/packages/meshes).
Supplementary data are available at Bioinformatics online.
1 Introduction
Characterization of the biological theme is an integral part for functional genomics studies. It provides insights for elucidating molecular signatures and mechanisms of complex biological phenomena. GO (Yu et al., 2010), DO (Yu et al., 2015b), KEGG (Yu et al., 2012) and Reactome (Yu and He, 2016) are the most widely used biological knowledge for the assessment of functional enrichment in biomedical science. Terms of GO and DO are organized as a directed acyclic graph, which have laid a foundation for computing semantic similarities among genes. Investigation of functional enrichment helps exploring biological meanings and unknown functional associations. GO and DO are the major resources for mining the biological knowledge to predict the functional associations based on semantic similarity measurement. Semantic similarity is widely used for genomic functional analysis, including protein-protein interaction, miRNA-mRNA interaction, cellular localization and motif analysis.
Medical Subject Headings (MeSH), which contains 19 categories, provides comprehensive biomedical vocabulary that are not covered by GO and other biomedical annotations, such as vocabularies in Anatomy, Chemicals and Drugs, Phenomena and Processes. In addition, the size of MeSH terms is approximately twice as large as that of GO, making it a good resource for interpreting biomedical data. Biological knowledge mining using MeSH annotation can enhance and enrich our functional interpretation. Previous study indicated that MeSH enables to extract broader meaning of genes compare to GO (Morota et al., 2015). Multiple packages have been developed for exploring functional associations and semantic analysis of genes using GO or DO, including GOSemSim (Yu et al., 2010), GOSim (Fröhlich et al., 2007), clusterProfiler (Yu et al., 2012) and DOSE (Yu et al., 2015b), but not MeSH. Although the R package, meshr (Tsuyuzaki et al., 2015), is capable of analyzing functional association using MeSH terms, this package only implemented over-representation analysis (ORA) and not allow users to perform gene set enrichment analysis (GSEA), which is more powerful for unveiling the perturbed pathways using expression profiles of the whole genome.
Here, I developed the meshes package that was designed directly for mining biological data based MeSH term. Both ORA and GSEA methods for the functional enrichment analysis as well as several methods for measuring semantic similarity among MeSH terms and gene products were implemented in meshes, which also provides several visualization methods for assisting result interpretation and producing publication-quality figures (Fig. 1).
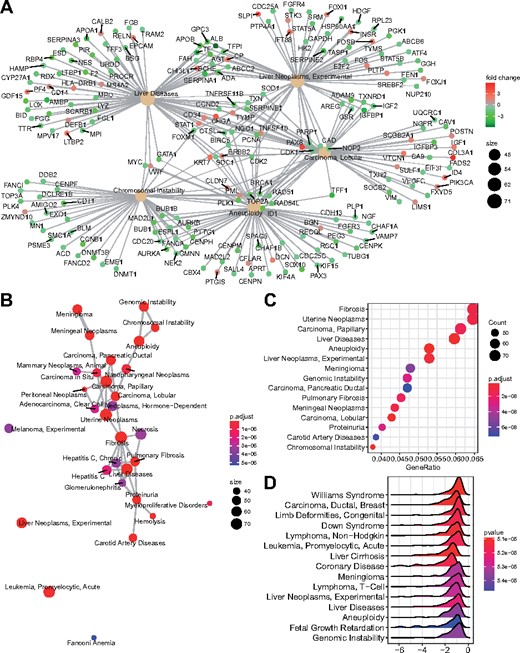
Visualization methods. (A) MeSH and gene association network; (B) Enrichment map; (C) Dot chart of most enriched terms; (D) Expression distribution of enriched gene sets
2 Implementation
The meshes package provides meshSim function to calculate semantic similarity among MeSH terms. It implements four information content based algorithms, namely ‘Resnik’, ‘Rel’, ‘Jiang’ and ‘Lin’ and one graph based algorithm, i.e. ‘Wang’. By mapping genes to MeSH terms, geneSim function computes semantic similarities among genes based on the similarity scores of annotated MeSH terms. Four combined strategies were implemented to aggregate semantic similarity scores. Computational details are described previously in GOSemSim (Yu et al., 2010) and can be referred to the online vignettes.
Gene-to-MeSH annotations are mainly generated by three methods using text mining (Gendoo), manual curation by NCBI (gene2pubmed) and sequence similarity using BLASTP search (RBBH). The meshes package supports more than 70 species listed in MeSHDb BiocView (https://bioconductor.org/packages/release/BiocViews.html#___MeSHDb). The enrichMeSH function implements hypergeometric model to investigate MeSH term associations of differential express genes and allows users to select an appropriate background (e.g. all genes quantified in an RNAseq experiment). The gseMeSH function provides GSEA algorithm to analyze the high-throughput data. Enrichment analyses are commonly used to verify functional associations and discover unanticipated functions.
The functionalities of meshes can be enhanced by other R packages, especially with in-house packages ChIPseeker (Yu et al., 2015a) and clusterProfiler (Yu et al., 2012). ChIPseeker provides a seq2gene function to link genomic regions to genes by many-to-many mapping. It takes genes which are possibly cis-regulated into consideration, such as host genes, promoter regions and flanking genes. The two-step approach that links genome-wide regions of interest (ROIs) to coding genes followed by enrichment analysis at gene level, enables the exploration of functional impact of genomic regions. The enrichment functions can be directly used in clusterProfiler to compare functional profiles for different conditions and/or at different time points using MeSH annotations.
3 Conclusion
The meshes package was developed as an R package and released within Bioconductor project. It provides five algorithms for semantic similarity measurement and two different approaches for enrichment analyses. More than 70 species were supported in this package to investigate functional associations of genomic data as well as knowledge mining based on MeSH term semantic relations. It fits the R ecosystem and works seamlessly with ChIPseeker to analyze NGS data and clusterProfiler for comparing different datasets. Moreover, meshes provides users with several visualization methods to produce customizable, high quality of figures to improve the ability of result interpretation. R scripts to generate Figure 1 were presented in Supplementary File. Details about the visualization methods can be found in the package’s online documentation.
Funding
None.
Conflict of Interest: none declared.
References


{kind=link}