





Abstract
The application of next-generation sequencing in research and particularly in clinical routine requires valid variant calling results. However, evaluation of several commonly used tools has pointed out that not a single tool meets this requirement. False positive as well as false negative calls necessitate additional experiments and extensive manual work. Intelligent combination and output filtration of different tools could significantly improve the current situation.
We developed appreci8, an automatic variant calling pipeline for calling single nucleotide variants and short indels by combining and filtering the output of eight open-source variant calling tools, based on a novel artifact- and polymorphism score. Appreci8 was trained on two data sets from patients with myelodysplastic syndrome, covering 165 Illumina samples. Subsequently, appreci8’s performance was tested on five independent data sets, covering 513 samples. Variation in sequencing platform, target region and disease entity was considered. All calls were validated by re-sequencing on the same platform, a different platform or expert-based review. Sensitivity of appreci8 ranged between 0.93 and 1.00, while positive predictive value ranged between 0.65 and 1.00. In all cases, appreci8 showed superior performance compared to any evaluated alternative approach.
Appreci8 is freely available at https://hub.docker.com/r/wwuimi/appreci8/. Sequencing data (BAM files) of the 678 patients analyzed with appreci8 have been deposited into the NCBI Sequence Read Archive (BioProjectID: 388411; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA388411).
Supplementary data are available at Bioinformatics online.
1 Introduction
Precision medicine is on its way to revolutionizing patient care. Individual therapeutic strategies are increasingly applied to provide every patient with the most suitable treatment. An important aspect for realizing personalized medicine with respect to genetically related diseases, including cancer, is the correct determination and interpretation of mutations (Ashley, 2016; Dey et al., 2017). In the course of the last years, this is increasingly done by next-generation sequencing (NGS) (Park et al., 2013).
Different from Sanger sequencing (Sanger et al., 1977), NGS provides a solution for detecting variants with variant allele frequencies (VAFs) below 20% (Mohamed et al., 2014). Furthermore, sequencing can be performed consuming only a fraction of time and costs (Loman et al., 2012), which enables the analysis of selected target regions as well as a patient’s whole exome or even whole genome.
When using NGS it is essential to be able to rely on variant calling results that are valid. Ideally, the analysis pipeline applied in research and particularly also in clinical routine has to feature both high sensitivity and high positive predictive value (PPV). However, all NGS platforms more or less suffer from systematic as well as random sequencing errors (Bragg et al., 2013; Hoffman et al., 2009; Liu et al., 2012; Yeo et al., 2014). Previously, we performed re-sequencing experiments involving several patients with myelodysplastic syndrome (MDS) that were sequenced on Illumina NextSeq, Ion Torrent PGM and Roche 454 platforms. These experiments revealed considerable differences in the number of true variants and artifacts reported per sample (Sandmann et al., 2017). These differences could be observed when comparing different sequencing platforms as well as when comparing two runs on the same platform.
The analysis of two Illumina data sets (HiSeq and NextSeq), covering altogether more than 150 patients with MDS indicated that additional differences in variant calling results can be expected when considering different variant calling tools (Sandmann et al., 2017). We considered all currently available open-source variant calling tools for NGS data. However, only 8 out of 43 tools were applicable on our sets of non-matched targeted sequencing data. Evaluation of these eight tools revealed that not a single tool succeeded in detecting all mutations present in the two data sets. Furthermore, no tool showed sensitivity and PPV for both data sets. Our observations are conform to the results of other studies comparing variant calling tools (Cornish and Guda, 2015; Hwang et al., 2015; Zook et al., 2014).
These studies point out the necessity for a variant calling pipeline that is able to detect variants with both high sensitivity and high PPV—even at low allelic frequencies. Additionally, the pipeline’s performance should be independent of the analyzed data set, not involve re-calibration with new training data in case of new experiments and not include validation of each variant call by Sanger sequencing as proposed for current pipelines by Mu et al. (2016). Furthermore, application should be possible even in the absence of normal controls, which is a common scenario as pointed out by Kalatskaya et al. (2017).
In this paper, we present ‘appreci8’—a Pipeline for PREcise variant Calling Integrating 8 tools. The pipeline automatically performs variant calling of single nucleotide variants (SNVs) and short indels integrating eight open-source variant calling tools. The calls are automatically normalized, combined and filtered on the basis of a novel artifact- and polymorphism score. The scores categorize a variant as either likely pathogenic mutation, polymorphism or artifact. Our tool is applicable to any type of NGS data.
To train our pipeline, we analyzed two sets of non-matched targeted sequencing data, covering 165 MDS patients sequenced on Illumina HiSeq, resp. Illumina NextSeq. An intersecting target region of 42 322 bp was considered. Performance of our pipeline was tested analyzing five independent sets of targeted sequencing data, differing from the training sets in varying degrees [sequencing platforms: Illumina HiSeq, HiScanSQ, NextSeq and Roche 454; target region: 42 322–958 547 bp; disease entity: MDS and acute myeloid leukemia (AML)]. Appreci8’s ability to separate true variants from artifacts with allelic frequencies down to 1% was evaluated. We compared our pipeline’s performance to every individual tools’ performance, all possible combined approaches and an alternative version of our pipeline.
2 Materials and methods
2.1 Variant calling pipeline
Appreci8 is a completely automatic pipeline for performing SNV and indel calling. An overview of the pipeline is provided in Figure 1.
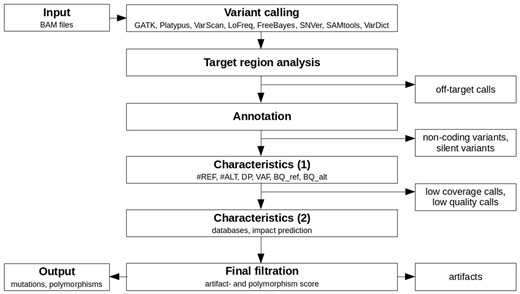
Overview of the analysis performed by appreci8
BAM files containing the raw aligned reads per sample form the input for our variant calling pipeline (see Supplementary Section 3 for information on sequence alignment). Variant calling is automatically performed on eight different tools: GATK 3.3-0 HaplotypeCaller (DePristo et al., 2011), Platypus 0.8.1 (Rimmer et al., 2014), VarScan 2.3.9 (Koboldt et al., 2012), LoFreq 2.1.2 (Wilm et al., 2012), FreeBayes 1.0.2 (Garrison and Marth, 2012), SNVer 0.5.3 (Wei et al., 2011), SAMtools 1.3 (Li et al., 2009) and VarDict (Lai et al., 2016). For each caller, the default recommended options are used. The only exception is the VAF threshold in case of FreeBayes and SNVer, which is lowered to 0.01 (default 0.20, resp. 0.25).
The resulting raw output per caller is filtered to remove all off-target calls. Subsequently, the remaining calls are combined (see Supplementary Section 4) and annotated using SnpEff (Cingolani et al., 2012). The user can choose between an annotation using ENSEMBL (Aken et al., 2016) or RefSeq (O’Leary et al., 2016). Furthermore, it is optional to report the annotation of all possible transcripts or just the annotation for selected transcripts. For our training and evaluation of appreci8, we removed all calls that are according to SnpEff located in the 3′-UTR, 5′-UTR, downstream, upstream, intron, intergenic, intragenic, protein–protein contact and in the splice site region (intron_variant+splice_region_variant). Furthermore, silent mutations were removed. By concentrating on coding, non-synonymous variants, we focus our analysis on those variants that are best characterized with respect to biological truth for all data sets considered. However, this filtration is not fixed, but can be adjusted.
For all remaining calls, appreci8 determines a first set of characteristics: the number of reference reads (#REF), the number of alternate reads (#ALT), the depth (DP) and the VAF. These characteristics are determined for all reads and for the forward- and reverse reads separately. Furthermore, the mean base quality (PHRED value) for the reference- () and alternate allele () are determined. As some of the tools apply specific steps of local realignment, all parameters are determined on the basis of the raw alignment data that have already been used for variant calling. Assuming that a decision on a call—whether or not it is true—is only possible in case of sufficient coverage, we remove all calls with number of alternate reads () <20, depth (DP) <50 or . Furthermore, we remove all calls with or (for details see Supplementary Section 5.1). All parameters may be easily adjusted depending on the sequencing data that are analyzed, e.g. in case of low-coverage whole-genome sequencing (WGS) data.
Finally, a second set of characteristics is determined for the remaining calls. These include the results of an automatic check of the databases ESP6500 (http://evs.gs.washington.edu/EVS/), 1000 Genomes (The 1000 Genomes Project Consortium, 2015), dbSNP (Sherry et al., 2001) (build 138 and build 138 excluding sites after 129), ExAC (Lek et al., 2016), Cosmic (Forbes et al., 2015) (CodingMuts, NonCodingVariants, CompleteExport and CompleteExport.fail, 17.02.2016) and ClinVar (Landrum et al., 2016) (common and clinical, 03.02.2016; common no known medical impact, 03.02.2016). Additionally, Provean 1.1.5 (Choi et al., 2012) is used to determine the influence of every variant on the corresponding protein.
Integrating all information characterizing a call, an artifact score—separating true from false positive calls—is calculated. Furthermore, a polymorphism score—identifying likely polymorphisms—is calculated. The general principle of filtration with appreci8 based on these two scores is displayed in Figure 2.
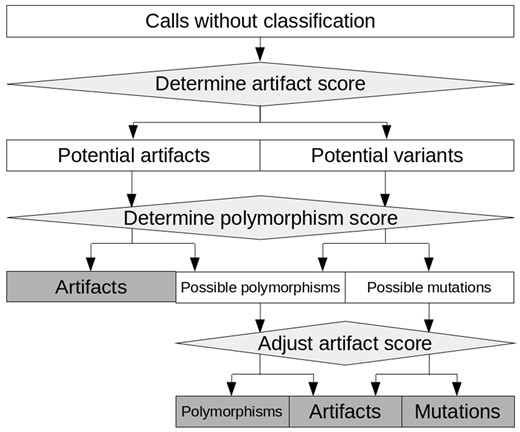
General principle of filtration with appreci8. Calls are classified as ‘Mutations’, ‘Polymorphism’ or ‘Artifact’ on the basis of an artifact- and a polymorphism score
The artifact score separates all calls into two initial categories: ‘Potential variants’ and ‘Potential artifacts’ (see Supplementary Fig. S3 for details). Subsequently, the polymorphism score is evaluated. It allows for separating ‘Possible mutations’ from ‘Possible polymorphisms’. However, for the final decision on these calls, the artifact score is reconsidered. It is adjusted on the basis of the polymorphism score as well as call characteristics. This enables the final classification of ‘Possible mutations’ and ‘Possible polymorphisms’.
In addition to separating mutations from polymorphisms, the polymorphism score enables the identification of ‘Polymorphisms’ in the initial set of ‘Potential artifacts’ (see Supplementary Fig. S4 for details). As some polymorphisms feature characteristics that are typical for artifacts, these calls would be misclassified on the basis of the artifact score alone, but are correctly classified by the combination of both scores.
The optimal weighting and combination of the different call characteristics for the calculation of the artifact- and polymorphism score is determined using two training sets (see Supplementary Sections 5.2 and 5.3 for details). The performance of appreci8 is evaluated analyzing five independent test sets.
2.2 Data sets analyzed
To train our variant calling pipeline—appreci8—two well characterized sets of amplicon-based targeted sequencing data are investigated. Both data sets result from MDS patients, covering an intersecting target region of 42 322 bp (19 genes). ‘Training set 1’ comprises 54 samples sequenced on Illumina HiSeq, using HaloPlex for target enrichment. ‘Training set 2’ comprises 111 samples sequenced on Illumina NextSeq, using TruSight DNA Amplicon Sequencing Panel Library Prep Kit (see Table 1).
Main characteristics of the training- and test sets analyzed with appreci8
| Set | n | Sequencer | Disease | Target [bp] | Coverage | Background | ||
|---|---|---|---|---|---|---|---|---|
| all | Coding | >50x (%) | noise | |||||
| Training | 1 | 54 | Illumina HiSeq | MDS | 42 322 | 23 162 | 95 | |
| 2 | 111 | Illumina NextSeq | MDS | 42 322 | 23 162 | 97 | ||
| Test | 1 | 237 | Illumina HiSeq | MDS | 42 322 | 23 162 | 92 | |
| 2 | 46 | Illumina HiSeq | MDS | 42 322 | 23 162 | 93 | ||
| 3 | 89 | Roche 454 | MDS | 42 322 | 23 162 | 84 | ||
| 4 | 22 | Illumina NextSeq | AML | 125 459 | 78 866 | 99 | ||
| 5 | 119 | Illumina HiScanSQ | AML | 958 547 | 218 179 | 94 | ||
| Set | n | Sequencer | Disease | Target [bp] | Coverage | Background | ||
|---|---|---|---|---|---|---|---|---|
| all | Coding | >50x (%) | noise | |||||
| Training | 1 | 54 | Illumina HiSeq | MDS | 42 322 | 23 162 | 95 | |
| 2 | 111 | Illumina NextSeq | MDS | 42 322 | 23 162 | 97 | ||
| Test | 1 | 237 | Illumina HiSeq | MDS | 42 322 | 23 162 | 92 | |
| 2 | 46 | Illumina HiSeq | MDS | 42 322 | 23 162 | 93 | ||
| 3 | 89 | Roche 454 | MDS | 42 322 | 23 162 | 84 | ||
| 4 | 22 | Illumina NextSeq | AML | 125 459 | 78 866 | 99 | ||
| 5 | 119 | Illumina HiScanSQ | AML | 958 547 | 218 179 | 94 | ||
Main characteristics of the training- and test sets analyzed with appreci8
| Set | n | Sequencer | Disease | Target [bp] | Coverage | Background | ||
|---|---|---|---|---|---|---|---|---|
| all | Coding | >50x (%) | noise | |||||
| Training | 1 | 54 | Illumina HiSeq | MDS | 42 322 | 23 162 | 95 | |
| 2 | 111 | Illumina NextSeq | MDS | 42 322 | 23 162 | 97 | ||
| Test | 1 | 237 | Illumina HiSeq | MDS | 42 322 | 23 162 | 92 | |
| 2 | 46 | Illumina HiSeq | MDS | 42 322 | 23 162 | 93 | ||
| 3 | 89 | Roche 454 | MDS | 42 322 | 23 162 | 84 | ||
| 4 | 22 | Illumina NextSeq | AML | 125 459 | 78 866 | 99 | ||
| 5 | 119 | Illumina HiScanSQ | AML | 958 547 | 218 179 | 94 | ||
| Set | n | Sequencer | Disease | Target [bp] | Coverage | Background | ||
|---|---|---|---|---|---|---|---|---|
| all | Coding | >50x (%) | noise | |||||
| Training | 1 | 54 | Illumina HiSeq | MDS | 42 322 | 23 162 | 95 | |
| 2 | 111 | Illumina NextSeq | MDS | 42 322 | 23 162 | 97 | ||
| Test | 1 | 237 | Illumina HiSeq | MDS | 42 322 | 23 162 | 92 | |
| 2 | 46 | Illumina HiSeq | MDS | 42 322 | 23 162 | 93 | ||
| 3 | 89 | Roche 454 | MDS | 42 322 | 23 162 | 84 | ||
| 4 | 22 | Illumina NextSeq | AML | 125 459 | 78 866 | 99 | ||
| 5 | 119 | Illumina HiScanSQ | AML | 958 547 | 218 179 | 94 | ||
To test the performance of appreci8 on independent data, we consider five additional sets of amplicon-based targeted sequencing data: ‘Test set 1’ covers Illumina HiSeq sequencing data (using HaloPlex for target enrichment) from 237 MDS patients. ‘Test set 2’ covers Illumina HiSeq sequencing data (using HaloPlex for target enrichment) from 46 MDS patients. ‘Test set 3’ covers Roche 454 (Janitz, 2008) sequencing data (using GS FLX Titanium SV emPCR Kit Lib-A) from 89 MDS patients. In case of these three test sets the same target region is analyzed as in the two training sets. ‘Test set 4’ covers Illumina NextSeq sequencing data (using TruSight DNA Amplicon Sequencing Panel Library Prep Kit) from 22 patients with acute myeloid leukemia (AML). Different from the first three test sets, a larger target region comprising 125 459 bp is analyzed in this case. ‘Test set 5’ covers Illumina HiScanSQ sequencing data (using HaloPlex for target enrichment) from 119 patients with AML. Again, a larger target region comprising 958 547 bp is analyzed.
In case of all data sets, patient material was collected and analyzed in accordance with the relevant ethical guidelines and regulations. Informed consent was obtained from all subjects. Sequencing data (BAM files) of the 678 patients have been deposited into the NCBI Sequence Read Archive (BioProjectID: 388411; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA388411).
We decided to choose these data sets for training and testing, as they are all well characterized with respect to biological truth. Furthermore, the sets allowed us to investigate if appreci8’s performance is dependent on the sequencing technique, the target region and the disease that is considered.
2.3 Validation
For the initial training of appreci8 and its subsequent evaluation, we only consider data sets with validated mutations. Validation was achieved using three different approaches: (i) a selected set of calls (mutations, polymorphisms and artifacts) was validated using Sanger sequencing. However, as variants with a VAF below 20% are difficult to confirm with this sequencing technique, (ii) we re-analyzed a subset samples by the same or another technique as validation. Six samples were re-analyzed on Illumina NextSeq. Nine samples were re-analyzed on Ion Torrent PGM (Rothberg et al., 2011). Twenty-two samples were analyzed on Roche 454 and Illumina NextSeq. NPM1 mutations were validated by LightCycler (Roche, Mannheim, Germany) based melting curve analysis (Schnittger et al., 2005). As additional validation (iii), all calls reported in case of the two training sets were manually investigated by two independent experts. The variant-specific characteristics as well as the calls themselves in the IGV (Robinson et al., 2011) were considered. In case of the five test sets, all calls categorized as true were manually investigated. Furthermore, variant-specific characteristics of all calls categorized as polymorphisms and artifacts were investigated.
3 Results
A variant calling pipeline’s main task is successfully calling true variants with high sensitivity and automatically discarding artifacts. Variants themselves can be subdivided into benign variants that are present in the general population, i.e. germline single nucleotide polymorphisms (SNPs) and indel polymorphisms, and possibly pathogenic variants, i.e. SNVs and indels. In cancer, most pathogenic mutations are somatically acquired and tumor cell specific. While the correct classification of polymorphisms versus pathogenic mutations is in some cases straight forward, it may often prove to be challenging due to lack of germline material, variants of uncertain clinical significance or subclonal variants. When considering a complete ClinVar (Landrum et al., 2016) export (August 2, 2016), the list contains 130 097 variants in total. About 32.63% are classified as variants of ‘uncertain clinical significance’ (24.02%) or variants with information on clinical significance ‘not provided’ (8.61%). Only 9.66% are classified as ‘benign’, 20.31% are classified as ‘pathogenic’. For these reasons, we consider the automatic separation between artifacts and true variants as the main task of our pipeline. The automatic separation between benign and pathogenic variants is considered an add-on and is presented in the supplement (see Supplementary Section 10).
3.1 Training appreci8
To train our variant calling pipeline, we use two well characterized NGS data sets. Although both sets are derived from patients with the same disease—MDS—and the same target region is analyzed, they differ in the enrichment technologies and sequencing platforms. Therefore, we expect to find different characteristics for both data and also variant calls.
Supplementary Table S1 shows that both training sets differ considerably in their main data characteristics. While training set 2 features higher mean coverage, the set is also characterized by 16% more background noise compared to training set 1. This characteristic is expected to have negative influence on PPV (see Supplementary Section 9 for information on how background noise was calculated).
Variant calling results with respect to sensitivity and PPV are displayed in Figure 3 and summed up in Table 2 (for details see Supplementary Tables S3 and S4 and Supplementary Data S1 and S2). The performance of every single tool—GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools and VarDict—is compared to appreci8. Additionally, we evaluate two alternative approaches in detail: ‘8 tools’ considers all variants that have been reported by at least one out of eight tools and no further steps of filtration. ‘Single-appreci8’ is an experimental variant of our algorithm. Every sample is evaluated independently. Any information on other samples analyzed in the same run is disregarded (see Supplementary Sections 6 and 7 for details on the two alternative approaches). In all cases, we only consider calls with sufficient coverage and , as we assume that validation of variants with lower VAFs or insufficient coverage is not feasible without further sequencing experiments.
Positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in training sets 1 and 2
| Approach | Training set 1 | Training set 2 | ||
|---|---|---|---|---|
| Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.85 | 0.82 | 0.71 |
| Platypus | 0.93 | 0.80 | 0.83 | 0.42 |
| VarScan | 0.89 | 0.97 | 0.47 | 0.73 |
| LoFreq | 0.91 | 0.35 | 0.78 | 0.23 |
| FreeBayes | 1.00 | 0.03 | 0.99 | 0.02 |
| SNVer | 0.93 | 0.92 | 0.55 | 0.07 |
| SAMtools | 0.85 | 0.87 | 0.64 | 0.77 |
| VarDict | 0.97 | 0.96 | 0.94 | 0.15 |
| 8 tools | 1.00 | 0.03 | 1.00 | 0.02 |
| single-appreci8 | 0.98 | 0.98 | 0.99 | 0.35 |
| appreci8 | 0.98 | 0.99 | 0.98 | 0.94 |
| Approach | Training set 1 | Training set 2 | ||
|---|---|---|---|---|
| Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.85 | 0.82 | 0.71 |
| Platypus | 0.93 | 0.80 | 0.83 | 0.42 |
| VarScan | 0.89 | 0.97 | 0.47 | 0.73 |
| LoFreq | 0.91 | 0.35 | 0.78 | 0.23 |
| FreeBayes | 1.00 | 0.03 | 0.99 | 0.02 |
| SNVer | 0.93 | 0.92 | 0.55 | 0.07 |
| SAMtools | 0.85 | 0.87 | 0.64 | 0.77 |
| VarDict | 0.97 | 0.96 | 0.94 | 0.15 |
| 8 tools | 1.00 | 0.03 | 1.00 | 0.02 |
| single-appreci8 | 0.98 | 0.98 | 0.99 | 0.35 |
| appreci8 | 0.98 | 0.99 | 0.98 | 0.94 |
Positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in training sets 1 and 2
| Approach | Training set 1 | Training set 2 | ||
|---|---|---|---|---|
| Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.85 | 0.82 | 0.71 |
| Platypus | 0.93 | 0.80 | 0.83 | 0.42 |
| VarScan | 0.89 | 0.97 | 0.47 | 0.73 |
| LoFreq | 0.91 | 0.35 | 0.78 | 0.23 |
| FreeBayes | 1.00 | 0.03 | 0.99 | 0.02 |
| SNVer | 0.93 | 0.92 | 0.55 | 0.07 |
| SAMtools | 0.85 | 0.87 | 0.64 | 0.77 |
| VarDict | 0.97 | 0.96 | 0.94 | 0.15 |
| 8 tools | 1.00 | 0.03 | 1.00 | 0.02 |
| single-appreci8 | 0.98 | 0.98 | 0.99 | 0.35 |
| appreci8 | 0.98 | 0.99 | 0.98 | 0.94 |
| Approach | Training set 1 | Training set 2 | ||
|---|---|---|---|---|
| Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.85 | 0.82 | 0.71 |
| Platypus | 0.93 | 0.80 | 0.83 | 0.42 |
| VarScan | 0.89 | 0.97 | 0.47 | 0.73 |
| LoFreq | 0.91 | 0.35 | 0.78 | 0.23 |
| FreeBayes | 1.00 | 0.03 | 0.99 | 0.02 |
| SNVer | 0.93 | 0.92 | 0.55 | 0.07 |
| SAMtools | 0.85 | 0.87 | 0.64 | 0.77 |
| VarDict | 0.97 | 0.96 | 0.94 | 0.15 |
| 8 tools | 1.00 | 0.03 | 1.00 | 0.02 |
| single-appreci8 | 0.98 | 0.98 | 0.99 | 0.35 |
| appreci8 | 0.98 | 0.99 | 0.98 | 0.94 |

Relation between positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in training sets 1 and 2
Figure 3 illustrates the data-dependent performance of the eight individual variant calling tools. While all tools succeed in calling variants with sensitivity above 0.80 in case of training set 1, only four tools—GATK, Platypus, FreeBayes and VarDict do so in case of training set 2. However, when aiming for sensitivity of at least 0.95, which appears to be a more apt threshold for usage of NGS in clinical routine, only two tools—FreeBayes (0.99) and VarDict (0.97)—succeed in case of training set 1 and only one tool—FreeBayes (0.99)—succeeds in case of training set 2.
While sensitivity is an essential aspect of any variant calling pipeline, so is PPV. False positive mutations can have serious consequences for the treatment of a patient. Furthermore, using a tool that reports thousands of calls per patient increases the risk of overlooking the actual true mutations among the many artifacts.
Regarding training set 1, six out of eight tools feature PPV above 0.80 (VarDict performs best with PPV = 0.96). FreeBayes, however, shows the lowest PPV (0.03). The tool reports 290 true variants and 8040 artifacts. Regarding training set 2, not a single tool reaches a value of 0.80 or above. FreeBayes reports 627 out of 631 true variants and 40,159 artifacts (PPV = 0.02).
When combining the output of all tools, we succeed in calling all variants present in both training sets (sens = 1.00). This is not possible with any of the considered individual tools. However, as PPV is 0.03 in case of training set 1 and 0.02 in case of training set 2, the need for filtration is obvious. Application of our appreci8 pipeline leads to a considerable increase in PPV—to values higher than any of the individual tools—while sensitivity is only marginally reduced. Comparing single-appreci8 to appreci8, only minor differences can be observed in case of training set 1. As regards training set 2, the application of appreci8 leads to a considerable improvement in the results (PPV = 0.35 versus PPV = 0.94). Evaluation of the artifact- and polymorphism score removes 1% of the artifacts in training set 1 and 21% in training set 2.
3.2 Testing appreci8
To test appreci8, we consider five independent, well characterized data sets (for data characteristics see Supplementary Table S2). Variant calling results with respect to sensitivity and PPV are displayed in Figure 4 and Table 3 (for details see Supplementary Tables S5–S9 and Supplementary Data S3–S7).
Positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in test sets 1–5
| Approach | Test set 1 | Test set 2 | Test set 3 | Test set 4 | Test set 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.82 | 0.90 | 0.73 | 0.95 | 0.31 | 0.86 | 0.73 | 0.92 | 0.94 |
| Platypus | 0.93 | 0.81 | 0.93 | 0.83 | 0.97 | 0.13 | 0.91 | 0.30 | 0.90 | 0.94 |
| VarScan | 0.84 | 0.84 | 0.82 | 0.73 | 0.91 | 0.51 | 0.73 | 0.55 | 0.71 | 0.94 |
| LoFreq | 0.98 | 0.29 | 0.98 | 0.71 | 0.94 | 0.31 | 0.73 | 0.01 | 0.79 | 0.88 |
| FreeBayes | 0.99 | 0.02 | 1.00 | 0.01 | 0.99 | 0.07 | 0.95 | 0.01 | 0.96 | 0.25 |
| SNVer | 0.91 | 0.91 | 0.94 | 0.81 | 0.97 | 0.10 | 0.64 | 0.04 | 0.73 | 0.98 |
| SAMtools | 0.82 | 0.83 | 0.81 | 0.75 | 0.93 | 0.68 | 0.70 | 0.53 | 0.86 | 0.99 |
| VarDict | 0.96 | 0.78 | 0.99 | 0.30 | 0.99 | 0.25 | 0.91 | 0.09 | 0.97 | 0.83 |
| 8 tools | 1.00 | 0.02 | 1.00 | 0.01 | 0.99 | 0.05 | 0.99 | 0.01 | 1.00 | 0.25 |
| single-appreci8 | 0.99 | 0.88 | 1.00 | 0.76 | 0.99 | 0.36 | 0.93 | 0.19 | 0.96 | 0.97 |
| appreci8 | 0.98 | 0.99 | 1.00 | 0.99 | 0.99 | 0.76 | 0.93 | 0.65 | 1.00 | 1.00 |
| Approach | Test set 1 | Test set 2 | Test set 3 | Test set 4 | Test set 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.82 | 0.90 | 0.73 | 0.95 | 0.31 | 0.86 | 0.73 | 0.92 | 0.94 |
| Platypus | 0.93 | 0.81 | 0.93 | 0.83 | 0.97 | 0.13 | 0.91 | 0.30 | 0.90 | 0.94 |
| VarScan | 0.84 | 0.84 | 0.82 | 0.73 | 0.91 | 0.51 | 0.73 | 0.55 | 0.71 | 0.94 |
| LoFreq | 0.98 | 0.29 | 0.98 | 0.71 | 0.94 | 0.31 | 0.73 | 0.01 | 0.79 | 0.88 |
| FreeBayes | 0.99 | 0.02 | 1.00 | 0.01 | 0.99 | 0.07 | 0.95 | 0.01 | 0.96 | 0.25 |
| SNVer | 0.91 | 0.91 | 0.94 | 0.81 | 0.97 | 0.10 | 0.64 | 0.04 | 0.73 | 0.98 |
| SAMtools | 0.82 | 0.83 | 0.81 | 0.75 | 0.93 | 0.68 | 0.70 | 0.53 | 0.86 | 0.99 |
| VarDict | 0.96 | 0.78 | 0.99 | 0.30 | 0.99 | 0.25 | 0.91 | 0.09 | 0.97 | 0.83 |
| 8 tools | 1.00 | 0.02 | 1.00 | 0.01 | 0.99 | 0.05 | 0.99 | 0.01 | 1.00 | 0.25 |
| single-appreci8 | 0.99 | 0.88 | 1.00 | 0.76 | 0.99 | 0.36 | 0.93 | 0.19 | 0.96 | 0.97 |
| appreci8 | 0.98 | 0.99 | 1.00 | 0.99 | 0.99 | 0.76 | 0.93 | 0.65 | 1.00 | 1.00 |
Positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in test sets 1–5
| Approach | Test set 1 | Test set 2 | Test set 3 | Test set 4 | Test set 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.82 | 0.90 | 0.73 | 0.95 | 0.31 | 0.86 | 0.73 | 0.92 | 0.94 |
| Platypus | 0.93 | 0.81 | 0.93 | 0.83 | 0.97 | 0.13 | 0.91 | 0.30 | 0.90 | 0.94 |
| VarScan | 0.84 | 0.84 | 0.82 | 0.73 | 0.91 | 0.51 | 0.73 | 0.55 | 0.71 | 0.94 |
| LoFreq | 0.98 | 0.29 | 0.98 | 0.71 | 0.94 | 0.31 | 0.73 | 0.01 | 0.79 | 0.88 |
| FreeBayes | 0.99 | 0.02 | 1.00 | 0.01 | 0.99 | 0.07 | 0.95 | 0.01 | 0.96 | 0.25 |
| SNVer | 0.91 | 0.91 | 0.94 | 0.81 | 0.97 | 0.10 | 0.64 | 0.04 | 0.73 | 0.98 |
| SAMtools | 0.82 | 0.83 | 0.81 | 0.75 | 0.93 | 0.68 | 0.70 | 0.53 | 0.86 | 0.99 |
| VarDict | 0.96 | 0.78 | 0.99 | 0.30 | 0.99 | 0.25 | 0.91 | 0.09 | 0.97 | 0.83 |
| 8 tools | 1.00 | 0.02 | 1.00 | 0.01 | 0.99 | 0.05 | 0.99 | 0.01 | 1.00 | 0.25 |
| single-appreci8 | 0.99 | 0.88 | 1.00 | 0.76 | 0.99 | 0.36 | 0.93 | 0.19 | 0.96 | 0.97 |
| appreci8 | 0.98 | 0.99 | 1.00 | 0.99 | 0.99 | 0.76 | 0.93 | 0.65 | 1.00 | 1.00 |
| Approach | Test set 1 | Test set 2 | Test set 3 | Test set 4 | Test set 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | Sens | PPV | |
| GATK | 0.92 | 0.82 | 0.90 | 0.73 | 0.95 | 0.31 | 0.86 | 0.73 | 0.92 | 0.94 |
| Platypus | 0.93 | 0.81 | 0.93 | 0.83 | 0.97 | 0.13 | 0.91 | 0.30 | 0.90 | 0.94 |
| VarScan | 0.84 | 0.84 | 0.82 | 0.73 | 0.91 | 0.51 | 0.73 | 0.55 | 0.71 | 0.94 |
| LoFreq | 0.98 | 0.29 | 0.98 | 0.71 | 0.94 | 0.31 | 0.73 | 0.01 | 0.79 | 0.88 |
| FreeBayes | 0.99 | 0.02 | 1.00 | 0.01 | 0.99 | 0.07 | 0.95 | 0.01 | 0.96 | 0.25 |
| SNVer | 0.91 | 0.91 | 0.94 | 0.81 | 0.97 | 0.10 | 0.64 | 0.04 | 0.73 | 0.98 |
| SAMtools | 0.82 | 0.83 | 0.81 | 0.75 | 0.93 | 0.68 | 0.70 | 0.53 | 0.86 | 0.99 |
| VarDict | 0.96 | 0.78 | 0.99 | 0.30 | 0.99 | 0.25 | 0.91 | 0.09 | 0.97 | 0.83 |
| 8 tools | 1.00 | 0.02 | 1.00 | 0.01 | 0.99 | 0.05 | 0.99 | 0.01 | 1.00 | 0.25 |
| single-appreci8 | 0.99 | 0.88 | 1.00 | 0.76 | 0.99 | 0.36 | 0.93 | 0.19 | 0.96 | 0.97 |
| appreci8 | 0.98 | 0.99 | 1.00 | 0.99 | 0.99 | 0.76 | 0.93 | 0.65 | 1.00 | 1.00 |
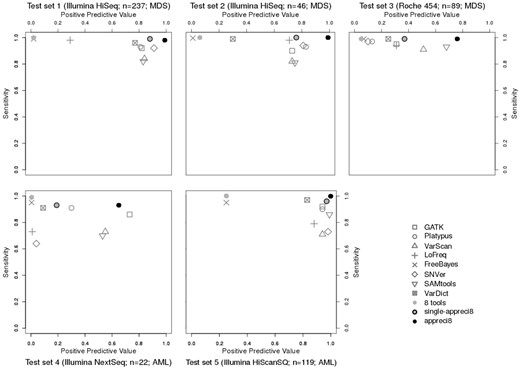
Relation between positive predictive value and sensitivity in case of GATK, Platypus, VarScan, LoFreq, FreeBayes, SNVer, SAMtools, VarDict, the combined output of all tools (eight tools), single-appreci8 and appreci8 in test sets 1–5
Test sets 1 and 2 result from the same sequencing platform as training set 1. Furthermore, the same target region and disease is considered as is the case for both training sets. Therefore, we expect all tools as well as our pipeline to show results comparable to training set 1.
When comparing Figures 1 and 2, it is clear that the results are indeed comparable. Sensitivity of the individual variant calling tools is above 0.80. FreeBayes, VarDict and LoFreq succeed in calling variants with sensitivity above 0.95 (test set 1: FreeBayes: 0.99, VarDict: 0.96, LoFreq: 0.98; test set 2: FreeBayes: 0.99, VarDict: 0.99, LoFreq: 0.98). Six out of eight tools feature PPV above or close to 0.80. Again, FreeBayes shows the lowest PPV (test set 1: 0.02; test set 2: 0.01). In contrast to training set 1, VarDict’s PPV is only 0.78 in test set 1 and 0.30 in test set 2.
The combined output of all tools leads to sens = 1.00 in case of both test sets. PPV = 0.02 for test set 1 and PPV = 0.01 for test set 2. Application of single-appreci8 leads to a minor decrease in sensitivity and to a considerable increase in PPV. Both test sets show a further clear improvement of the results when applying appreci8 in its actual functionality (test set 1: sens = 0.98, PPV = 0.99; test set 2: sens = 1.00, PPV = 0.99). Evaluation of the artifact- and polymorphism score removes 2% of the artifacts in test set 1 and 9% in test set 2.
To test appreci8’s robustness with respect to variation in the sequencing technique, we analyzed Roche 454 data (test set 3), although the pipeline was exclusively trained on Illumina data. Regarding the individual tools, sensitivity ranges between 0.91 and 0.99, while PPV ranges between 0.07 and 0.68. By combining the output of all variant calling tools, sensitivity increases to 0.99, while PPV is 0.05. Application of single-appreci8 leads to an improvement in the results. The overall best results can, however, be observed when applying appreci8 (sens = 0.99, PPV = 0.76). Remarkably, 97% of the artifacts are filtered because of their artifact- and polymorphism score.
To test appreci8’s robustness when considering a bigger target region and a different disease entity, we analyzed test sets 4 and 5. Both data sets result from patients with AML and cover a considerably bigger target region (test set 4: 125 459 bp, test set 5: 958 547 bp in comparison to 42 322 bp for the MDS training sets).
Test set 4 underlines the data-dependent performance of individual variant calling tools. Low sensitivity (ranging between 0.64 and 0.95) and low PPV (ranging between 0.01 and 0.73) is observed for the eight tools. Combining the output of all tools leads to sensitivity of 0.99 and a PPV of 0.01. Application of single-appreci8 leads to a minor decrease in sensitivity and an increase in PPV. Application of appreci8 leads to further improvement of the variant calling results (sens = 0.93, PPV = 0.65).
Considering test set 5, FreeBayes and VarDict feature again highest sensitivity of all individual variant calling tools (FreeBayes: 0.95, VarDict: 0.96, other tools: 0.71–0.91). Higher values of PPV can be observed in comparison to all previously analyzed data sets. Even FreeBayes features PPV of 0.25 (other tools: 0.81–0.99). The combined output of all tools leads to sens = 0.99 and PPV = 0.25. Application of single-appreci8 results in a minor decrease in sensitivity and to a considerable increase in PPV. Application of appreci8 leads to the best variant calling results that can be observed for this data set (sens = 0.99, PPV = 0.99).
Analyzing the influence of reoccurring variants, it can be observed that appreci8 performs equally well—detecting variants known from the training sets as well as unknown variants (see Supplementary Section 13).
4 Discussion
In the context of high-throughput research sequencing and personalized medicine, NGS provides a powerful tool. When considering variant calling results, it is therefore essential to be able to rely on a tool with stable and high sensitivity as well as high PPV. However, the analysis of seven data sets of non-matched amplicon-based targeted sequencing data, covering 678 samples from patients with hematological malignancies, shows that no individual tool meets these requirements.
We developed a pipeline, appreci8, that automatically combines and filters the variant calling results of eight different tools. Appreci8 succeeds in separating true calls from artifacts in all analyzed data sets with sensitivity ranging between 0.93 and 1.00 and PPV ranging between 0.65 and 1.00. Appreci8’s performance is in all cases superior to the best individual tool as well as to alternative combined approaches. Application of appreci8 on additional Illumina data sets (HiSeq, MiSeq and NextSeq), which were not part of this study (data available on request) as well as Ion Torrent data (see Supplementary Section 11), shows comparable results. Application of appreci8 on a public targeted sequencing data set (Sequence Read Archive, project PRJEB14077) shows comparable results as well (see Supplementary Section 12). Our results indicate that appreci8 is a pipeline that can generally be applied on NGS data, independent of the sequencing technique, the disease entity or the genes that are studied.
It should also be noted that appreci8 is able to automatically exclude polymorphisms, while all the other individual tools require manual filtration of polymorphisms or—in case of GATK—filtration on the basis of predefined polymorphism files.
It can be discussed, why we chose exactly this set of variant calling tools. Altogether, we used all available open-source tools that could be applied on our sets of non-matched targeted NGS data. However, performing variant calling with eight instead of one tool has negative influence on run-time if a high-performance server is not available. Furthermore, post-processing—which is inevitable—is time-consuming as well. We cannot exclude the possibility that seven or even less tools might also lead to acceptable results—dependent on the analyzed data set. However, for the calculation of the artifact- and the polymorphism score, the number of callers and even the specific callers that detect a variant are important characteristics. Decreasing the number of tools would thus have negative influence on appreci8’s overall performance.
Analysis of the overlapping calls, reported by two to eight tools, indicates that a mere combination of tools—even those with different variant detection algorithms—would not be beneficial (see Supplementary Section 8).
It can also be questioned, why we chose our set of 41 conditions that are evaluated to calculate the artifact- and the polymorphism score. On the one hand, our selection covers all classical characteristics that are considered when separating true from false positive calls, e.g. coverage, base quality and strand bias. On the other hand, we additionally consider novel characteristics, e.g. the number of tools calling a variant or the Provean score. Altogether, our categories represent the joined experience of biological and bioinformatical experts. Instead of being a black box, our algorithm aims at comprehensibly reproducing a biologist’s manual work when investigating a raw list of calls.
The weights we assigned to the different conditions were determined exploratively to optimize performance of appreci8 in case of the two training sets. The results we observe regarding the independent test sets are comparable. Still it is possible that another weighting or evaluation of the conditions might have led to even better results. Alternatively, we could have used decision tree learning or estimated a model, e.g. a generalized linear model. The parameters—the conditions in our case—and their weights would have been selected with the help of a model selection approach and an information criterion (Sandmann et al., 2017). However, the determined model or decision tree would be exclusively based on the two training sets. On the contrary, our approach additionally considers long-time experience of molecular biology experts.
Another approach would be to apply deep learning to estimate the best model, like Esteva et al. (2017) did. However, this approach has the disadvantage of being another black box. Manual adaptation of the parameters or their weighting based on e.g. new experience or updates of the data bases is not possible.
On purpose, we did not include any platform specific characteristics or filtration steps. Appreci8 was developed as a thoroughly automatic pipeline that does not require data-dependent re-calibration. Still—if desired—a user has the possibility to adjust thresholds, e.g. regarding coverage if low-coverage WGS instead of high-coverage targeted sequencing data is analyzed. Furthermore, we did not consider any tool-specific filtration steps or changes in the tool-specific parameters for variant calling. These might have significant influence on the different tools’ sensitivities and PPVs. However, GATK is the only tool that proposes precise thresholds for filtration. All the other tools do not. Additionally, no tool provides data-dependent recommended configurations for variant calling. Due to the high number of possible configurations, we decided to treat all callers equally, not apply any tool-specific filtration steps and stick to the default options for variant calling.
An essential aspect of our analysis is the correct classification of all variant calls. We did not validate every single call by re-sequencing the corresponding sample on the same or another platform. Instead, a majority of calls were validated by expert-based review. It could be argued that this approach may have led to mistakes. However, we were facing more than 180 000 calls in total, i.e. on average almost 270 calls per patient. By performing re-sequencing experiments in case of exemplary mutations, polymorphisms and artifacts, we showed that our classification was indeed correct. Furthermore, expert-based review involved evaluation of various databases, base qualities, coverage, allelic frequencies, the predicted effect on protein level and manual inspection using IGV, considering the sample in question and other samples in the same run. For these reasons, we estimate the risk of mistakes in the classification to be low and not to influence our overall results.
It can be observed that even when using appreci8, sensitivity and PPV are still lower than 1.00. Detailed analyses of the false positive- and false negative calls reveal that many feature an artifact score between −1 and 1 (threshold for true positive calls: −1). This observation suggests that additional analyses of calls with an artifact score close to the threshold could be useful. We are currently testing an approach evaluating the signal-to-noise ratio as described by Kockan et al. (2017) in combination with base qualities.
Regarding run-time, we are currently testing a speed-up version of our pipeline that improves the analysis of whole-exome sequencing and WGS data. Previous analyses have shown that it is not advisable to use the multi-threading modes of the variant calling tools (Sandmann et al., 2017).
While germline samples can already be analyzed with appreci8, automatic filtration of germline calls by matched sample analysis is not yet available. We are currently investigating an algorithm for automatic filtration of germline calls from matching, as well as pooled control samples with our appreci8 pipeline. As soon as this approach is available, it will be interesting to compare appreci8 with popular tools for matched sample analysis, e.g. MuTect2 (Cibulskis et al., 2013).
Considering the ongoing development in the field of variant calling, it appears useful to make appreci8 more flexible with respect to the variant calling tools that are considered. We are currently testing an extension of appreci8 that allows the user to select his own set of variant calling tools to consider. Tool versions and configurations used for variant calling will be user-definable. A graphical user interface will be provided to facilitate application of appreci8.
5 Conclusion
To consider variant calling results in research and in clinical routine, it is necessary to have a tool with stable, high sensitivity as well as high PPV. However, the analysis of seven data sets, covering 678 samples from patients with hematological malignancies, shows that no individual tool meets these requirements.
We developed a pipeline, appreci8, that combines and filters the variant calling results of eight different tools. Appreci8 succeeds in separating true calls from artifacts in all analyzed data sets with sensitivity ranging between 0.93 and 1.00 and PPV ranging between 0.65 and 1.00. Appreci8’s performance is in all cases superior to the best individual tool.
Acknowledgements
We thank the Genome Technology Center, Radboud university medical center, for performing the NextSeq500 sequencing runs. Sequencing of the HiSeq was performed at Sci-Life lab sequencing Facilities.
Funding
This work was supported by European Union grants Triage-MDS [ERA-Net TRANSCAN BMBF 01KT1401] and -Horizon2020 MDS-RIGHT [634789]; Deutsche Krebshilfe grant ‘Verbesserung der Diagnostik von Tumorerkrankungen durch neue DNA-Sequenzierverfahren und Algorithmen’ [110495]; collection of the HiSeq samples was performed by a grant from Swedish Cancer Society to Eva Hellström-Lindberg.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}