





Abstract
MicroRNAs (miRNAs) are small non-coding RNAs with the length of ∼22 nucleotides. miRNAs are involved in many biological processes including cancers. Recent studies show that long non-coding RNAs (lncRNAs) are emerging as miRNA sponges, playing important roles in cancer physiology and development. Despite accumulating appreciation of the importance of lncRNAs, the study of their complex functions is still in its preliminary stage. Based on the hypothesis of competing endogenous RNAs (ceRNAs), several computational methods have been proposed for investigating the competitive relationships between lncRNAs and miRNA target messenger RNAs (mRNAs). However, when the mRNAs are released from the control of miRNAs, it remains largely unknown as to how the sponge lncRNAs influence the expression levels of the endogenous miRNA targets.
We propose a novel method to construct lncRNA related miRNA sponge regulatory networks (LncmiRSRNs) by integrating matched lncRNA and mRNA expression profiles with clinical information and putative miRNA-target interactions. Using the method, we have constructed the LncmiRSRNs for four human cancers (glioblastoma multiforme, lung cancer, ovarian cancer and prostate cancer). Based on the networks, we discover that after being released from miRNA control, the target mRNAs are normally up-regulated by the sponge lncRNAs, and only a fraction of sponge lncRNA-mRNA regulatory relationships and hub lncRNAs are shared by the four cancers. Moreover, most sponge lncRNA-mRNA regulatory relationships show a rewired mode between different cancers, and a minority of sponge lncRNA-mRNA regulatory relationships conserved (appearing) in different cancers may act as a common pivot across cancers. Besides, differential and conserved hub lncRNAs may act as potential cancer drivers to influence the cancerous state in cancers. Functional enrichment and survival analysis indicate that the identified differential and conserved LncmiRSRN network modules work as functional units in biological processes, and can distinguish metastasis risks of cancers. Our analysis demonstrates the potential of integrating expression profiles, clinical information and miRNA-target interactions for investigating lncRNA regulatory mechanism.
LncmiRSRN is freely available (https://github.com/zhangjunpeng411/LncmiRSRN).
Supplementary data are available at Bioinformatics online.
1 Introduction
With the advance of next-generation sequencing technologies, non-coding RNAs (ncRNAs) as functional RNA molecules have challenged the traditional view of genome organization, with which genetic information is only stored in protein-coding genes (Evans et al., 2016). In recent years, long non-coding RNAs (lncRNAs), with longer than 200 nucleotides in length (Derrien et al., 2012), have attracted much attention from researchers in various fields as a major class of important ncRNAs. Accumulating evidence has revealed that lncRNAs are involved in a wide range of biological processes, such as gene transcription, post-transcriptional regulation, epigenetic regulation, and even human cancers (Cao, 2014; Chen et al., 2017; Wu et al., 2016).
Recently, the hypothesis on competing endogenous RNA (ceRNA) has been proposed (Salmena et al., 2011), and it is regarded as the ‘Rosetta Stone’ of a hidden RNA language. According to the hypothesis, a pool of different RNAs, including lncRNAs, pseudogenes, circular RNAs (circRNAs) and messenger RNAs (mRNAs) compete for the same pool of microRNAs (miRNAs), thereby regulating miRNA activity (Tay et al., 2014). miRNAs are small non-coding RNAs with the length of ∼22 nucleotides, and they function in RNA silencing and post-transcriptional regulation of gene expression. These ceRNAs are therefore also called miRNA ‘sponges’ or ‘decoys’. They act as molecular sponges to attract miRNAs for binding and competitively sequester them from their natural targets, and thus releasing target genes from the miRNAs’ control. As a representative of the several types of ceRNAs, lncRNAs, when acting as miRNA sponges, are associated with various human diseases, such as glioblastoma multiforme (Zhang et al., 2016a), lung cancer (Sun et al., 2016), ovarian cancer (Zhou et al., 2016) and prostate cancer (Zhang et al., 2017).
To study the system-level properties of lncRNAs as miRNA sponges in human cancers, several computational analysis methods (Conte et al., 2017; Du et al., 2016; Paci et al., 2014; Sui et al., 2016; Sumazin et al., 2011; Wang et al., 2015; Zhang et al., 2016b) have been presented to identify lncRNA related miRNA sponge networks.
Sumazin et al. (2011) proposed a miRNA activity modulator screening algorithm called Hermes to identify miRNA-mediated network of coding and non-coding RNA interactions, by analyzing matched miRNA and gene expression profiles in glioblastoma. The method utilizes mutual information and conditional mutual information to evaluate the statistical significance of each (RNA, miRNA, RNA) triplet. The constructed miRNA-mediated network of RNA-RNA interactions provides clues to the dysregulation of key mechanisms of pathogenesis, as well as to the regulation of normal cell physiology.
By estimating the so-called sensitivity correlation (the difference between Pearson and partial correlation coefficients) for each (lncRNA, miRNA, RNA) triplet.), Paci et al. (2014) and Conte et al. (2017) investigated the ability of lncRNAs to act as miRNA sponges by protecting mRNAs from miRNA repression. By dividing the breast tissues into tumor and normal breast samples, they built two types of miRNA-mediated interaction (MMI) networks: tumor and normal MMI networks, respectively. There is a marked rewiring in the lncRNA related miRNA sponge interactions between tumor and normal breast tissues, indicating an underlying role by the miRNA sponges (i.e. lncRNAs) as potential oncogenes or antioncogenes in cancer.
In addition, based on integrative analysis, Wang et al. (2015), Du et al. (2016) and Sui et al. (2016) investigated lncRNA related miRNA sponge networks in human cancer. The findings provide insights for better understanding the critical role of lncRNA-related sponge regulation in cancer. To understand the global regulation landscape and the characteristics of lncRNA related miRNA sponge crosstalk in cancers, Zhang et al. (2016b) integrated multidimensional molecule profiles of >5000 samples to systematically characterize lncRNA related miRNA sponge network across 12 major cancers. This study sheds light on the understanding of the molecular mechanism of tumorigenesis.
Although the above methods can be applied to investigate lncRNA related miRNA sponge networks, most of them rely on predicted miRNA-target interactions to generate candidate lncRNA-mRNA pairs. It is well known that different miRNA-target prediction programs use different techniques and metrics, which may cause inconsistent prediction results (Ekimler and Sahin, 2014). Moreover, these miRNA-target prediction algorithms produce many false positives and the number of biologically relevant miRNA target genes is largely overestimated (Pinzón et al., 2017), affecting the accuracy of the findings.
Additionally, miRNA sponges compete with mRNAs to attract miRNAs for binding and reducing the amount of miRNA transcripts. These miRNA sponges competitively sequester miRNAs from the target mRNAs, therefore releasing mRNAs from miRNAs’ control. When mRNAs are released from miRNAs’ control, an open question is how the expression levels of the released mRNAs are activated. One possible explanation is that the expression levels of the released mRNAs are typically activated by themselves, and can in principle be translated. In fact, the explanation is an implicit corollary of the ceRNA hypothesis. However, if the released mRNAs are still in an inactivated state, how are their expression levels activated? Previous studies (Faghihi et al., 2008,, 2010) have shown that lncRNAs could increase mRNA stability and thus regulate mRNA expression. Therefore, a possible interpretation is that the expression levels of these released mRNAs are activated by their competitive partners, e.g. sponge lncRNAs. In this paper, we hypothesize that lncRNAs as potential regulators activate the expression levels of the mRNAs released from miRNAs’ control. The aim of this paper is thus to investigate the regulatory relationships between the lncRNAs and the released mRNAs. This aim also differentiates our method from the existing methods, which are only aimed at identifying lncRNA related sponge networks.
To investigate how sponge lncRNAs influence the expression levels of the released mRNAs, it is necessary to uncover the regulatory mechanism of sponge lncRNAs. Existing methods only study crosstalk (indirect) or competitive relationships between sponge lncRNAs and mRNAs based on statistical correlations or associations whereas the regulatory relationships between sponge lncRNAs and mRNAs are indeed causal.
To address the above questions and limitations, in this work, we propose a causality-based computational method to identify lncRNA related miRNA Sponge Regulatory Network (LncmiRSRN). Firstly, to avoid inconsistent prediction results between different miRNA-target prediction algorithms, we integrate several well-known experimentally validated miRNA-target interaction databases as ground-truth to generate candidate sponge lncRNA-mRNA pairs. Furthermore, we assume that the expression levels of the released mRNAs are activated by their competitive partner lncRNAs in the sponge network. To find out if regulatory relationships exist between candidate sponge lncRNA and mRNA pairs, we use a causal inference method called intervention calculus when the DAG is absent (IDA) (Maathuis et al., 2009,, 2010), to estimate the causal effects of sponge lncRNAs on the mRNAs. To improve computation efficiency, we use the parallelized version of IDA called IDA_parallel (Le et al., 2016) to estimate the causal effects that a sponge lncRNA has on an endogenous miRNA target.
To validate the proposed LncmiRSRN method, we apply it to gene expression profiles and clinical information of four different cancer types: glioblastoma multiforme (GBM), lung squamous cell carcinoma (LSCC), ovarian cancer (OvCa) and prostate cancer (PrCa). We identify lncRNA related miRNA sponge regulatory networks of the four cancers and conduct functional analysis on them. The results show that the proposed method can help to elucidate sponge lncRNA related regulatory mechanisms in cancers.
2 Materials and methods
2.1 Matched lncRNA and mRNA expression profiles of human cancers
The genome-wide matched lncRNA and mRNA expression profiles of human cancers are obtained from (Du et al., 2013). By re-annotating the probes uniquely mapped to lncRNAs, Du et al. repurposed the publically available array-based data to extract lncRNA expression data. The lncRNAs and mRNAs without gene symbols in the repurposed microarray data are removed, and the unique expression value of replicate lncRNAs and mRNAs is obtained by taking the average of expression values of the replicates. As a result, we have the expression profiles of 9704 lncRNAs and 18 282 mRNAs in 451 GBM samples, 113 LSCC samples, 585 OvCa samples and 150 PrCa samples. The clinical information of the 451 GBM samples, 113 LSCC samples, 585 OvCa samples is from The Cancer Genome Atlas (TCGA) project (Weinstein et al., 2013), and the clinical information of the 150 PrCa samples is obtained from the Memorial Sloan-Kettering Cancer Center (MSKCC) prostate oncogenome project (Taylor et al., 2010).
2.2 Putative miRNA-target interactions
To collect putative miRNA-target interactions (including miRNA-mRNA and miRNA-lncRNA interactions), we integrate several well-known experimentally validated miRNA-target interaction databases. According to the catalog of experimental evidences in miRTarBase v7.0 (Chou et al., 2018) database, we also divide the experimental evidences of putative miRNA-target interactions into two types: strong and weak. For miRNA-mRNA interactions, we obtain the interactions with strong experimental evidences by integrating miRTarBase v7.0 (Chou et al., 2018) and TarBase v7.0 (Vlachos et al., 2015). Since the number of miRNA-lncRNA interactions with strong experimental evidences is very few, we combine the interactions with both strong and weak experimental evidences from NPInter v3.0 (Hao et al., 2016) and LncBase v2.0 (Paraskevopoulou et al., 2016). In total, we have collected 9318 and 17 3468 unique putative miRNA-mRNA and miRNA-lncRNA interactions, respectively.
2.3 Overview of LncmiRSRN
In this section, we will briefly describe the LncmiRSRN method for constructing a lncRNA related miRNA sponge regulatory network. As shown in Figure 1, the method includes the following steps:
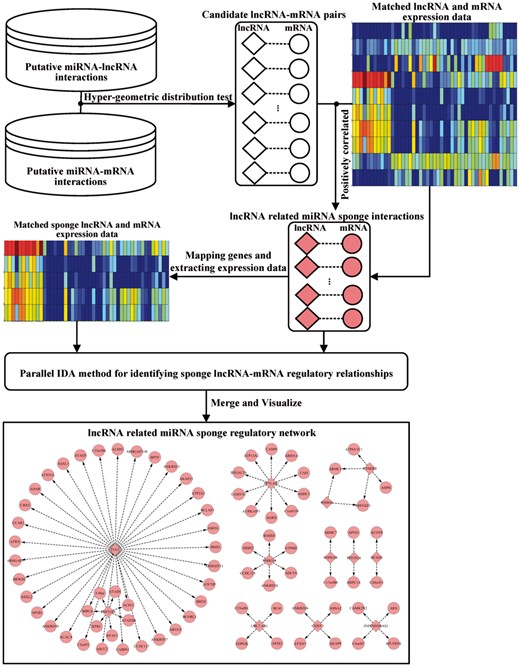
The workflow of LncmiRSRN. Firstly the putative miRNA-target interactions are used to generate candidate lncRNA-mRNA pairs. By using matched lncRNA and mRNA expression data, we select the candidate lncRNA related miRNA sponge interactions based on the correlations of the expression levels of the lncRNA-mRNA pairs. Then we identify sponge lncRNA-mRNA regulatory relationships with parallel IDA method. By merging these sponge lncRNA-mRNA regulatory relationships, we build the lncRNA related miRNA sponge regulatory network
Inferring lncRNA related miRNA sponge interactions. Given putative miRNA-target interactions, a list of candidate lncRNA-mRNA pairs which have significant sharing of miRNAs are identified. Each candidate lncRNA-mRNA pair with significant positive correlation is considered as a lncRNA related miRNA sponge interaction.
Estimating the causal effects of sponge lncRNAs on mRNAs. For the lncRNAs and mRNAs of the above identified sponge lncRNA-miRNA interaction pairs, we firstly extract matched sponge lncRNA and mRNA expression data. Then we apply parallel IDA to the matched lncRNA and mRNA expression data to calculate the causal effects of the sponge lncRNAs on the mRNAs.
Constructing lncRNA related miRNA sponge regulatory network. By using corPvalueFisher function of the R-package WGCNA (Langfelder and Horvath, 2008), we calculate the Fisher’s asymptotic P-values based on the causal effects of sponge lncRNAs on mRNAs to evaluate the strengths of sponge lncRNA-mRNA interactions. A lower P-value indicates a stronger sponge lncRNA-mRNA interaction. The sponge lncRNA-mRNA interactions with adjusted P-value < 0.05 (adjusted by BH method) are regarded as sponge lncRNA-mRNA regulatory relationships. By assembling these regulatory relationships, we form the lncRNA related miRNA sponge regulatory network.
2.4 Identifying lncRNA related miRNA sponge interactions
We mainly follow the two commonly used principles described below to identify lncRNA related miRNA sponge interactions.
Firstly, each candidate lncRNA-mRNA pair should have a significant sharing of miRNAs at sequence level. In this work, we require that each candidate lncRNA-mRNA pair share at least three miRNAs, and pass the significance test on the sharing with adjusted P-value < 0.01 (adjusted by BH method) using a hyper-geometric distribution test.
Secondly, the expression levels of each candidate lncRNA-mRNA pair are positively correlated. To identify lncRNA related miRNA sponge interactions, we compute the Pearson correlation coefficients of each candidate lncRNA-mRNA pair. All the candidate lncRNA-mRNA pairs with positive correlation coefficients and adjusted P-value < 0.01 (adjusted by BH method) are regarded as lncRNA related miRNA sponge interactions.
2.5 Constructing lncRNA related miRNA sponge regulatory networks
For the lncRNAs and mRNAs identified to possibly have lncRNA related miRNA sponge interactions, we extract their matched expression data. Then we identify sponge lncRNA-mRNA regulatory relationships based on the matched sponge lncRNA and mRNA expression data.
In this work, we apply IDA (Maathuis et al., 2009,, 2010) to the matched sponge lncRNA and mRNA expression data to identify the regulatory relationships in two steps: (i) considering the sponge lncRNAs and mRNAs as variables and learn the causal structure (links) among these variables from expression data and (ii) estimate the causal effects of sponge lncRNAs on mRNAs.
In step (i), we learn the causal structure from expression data using the PC algorithm (Spirtes et al., 2000), a well-known algorithm for causal structure learning based on conditional independence tests. To improve efficiency, we use the parallel implementation of PC in the R-package, ParallelPC (Le et al., 2016), when setting the significant level of the conditional independence tests, = 0.01. In this paper we assume that the regulatory relationships of the variables (sponge lncRNAs or mRNAs) can be represented using a directed acyclic graph (DAG), where a directed link A→B indicates that A is a regulator of B. DAGs have been commonly used to model gene regulatory relationships (Friedman, 2004; Friedman et al., 2000). Since different DAGs may correspond to the same conditional independence in a dataset. For example, A→B→C, A←B←C and A←B→C all show that A and C are conditional independent given B. The PC algorithm in this case outputs a CPDAG (completed partial DAG) of which some edges may be unidirectional. For this example, PC will output A-B-C to represents the equivalence class of DAGs, A→B→C, A←B←C and A←B→C. Additionally, to deal with high-dimensional expression data, an efficient conditional independent test, partial correlation test (Kalisch and Bühlmann, 2007) is used by the PC algorithm.
Given N variables or nodes, to determine if there is an edge between each pair of nodes based on conditional independence (CI) tests, in the worst case, the number of CI tests is N(N − 1)2N−2, which is intractable for large N. To tackle the challenge, the PC algorithm starts with a fully connected graph, it then removes an edge if a CI test returns true for the edge. As PC conducts CI tests in a level by level manner (starting with order zero CI tests) and each CI test is only conditioned on the neighbours of the two nodes being tested, when the underlying true causal DAG is sparse and it is possible to detect CI at lower level, the number of neighbours of a node will drop quickly when the level of CI tests goes up. So in practice, the number of CI tests conducted by PC is much smaller than that in the worst case. To further improve efficiency, in this paper, as mentioned earlier, we use the parallel implementation of PC in the R-package, ParallelPC (Le et al., 2016).
In step (ii), based on the learnt causal structure and the gene expression data, for each sponge lncRNA, we estimate its causal effect on all mRNAs by following the IDA method. Details of IDA are out of the scope of this paper, and we refer readers to (Le et al., 2016; Maathuis et al., 2009,, 2010) for more information.
The estimated causal effects can be positive or negative. We evaluate the strengths of identified sponge lncRNA-mRNA regulatory relationship using the Fisher’s asymptotic P-values. A sponge lncRNA-mRNA interaction with adjusted P-value < 0.05 (adjusted by BH method) is regarded as a sponge lncRNA-mRNA regulatory relationship. By assembling all the regulatory relationships, we obtain the lncRNA related miRNA sponge regulatory network (LncmiRSRN).
2.6 Topological properties of the LncmiRSRNs
The R-package igraph (Csardi and Nepusz, 2006) is used to analyze the topological properties of the LncmiRSRNs of the four cancers. For each node in a LncmiRSRN, its degree is defined as the number of edges connected with it. If the node degree in the LncmiRSRN obeys a power law model, the network is regarded as scale free, which is one of the most important metrics of true biological networks (Barabási and Oltvai, 2004). Previous studies have reported that hub genes with higher degrees tend to be essential, and nearly 20% of the nodes in a biological network are regarded as essential nodes (Hahn and Kern, 2005; Song and Singh, 2013). Therefore, in this work, we select the top 20% of lncRNAs with the highest degrees in the LncmiRSRN as the hub lncRNAs.
2.7 Survival and enrichment analysis for LncmiRSRN network modules
Before survival and enrichment analysis of each of the LncmiRSRNs, we firstly generate LncmiRSRN modules. In this work, we use the Markov Clustering Algorithm (MCL) (Enright et al., 2002) implemented in the R-package ProNet (Wu and Xia, 2015) to identify LncmiRSRN modules. For each module, the numbers of sponge lncRNAs and mRNAs are at least two, respectively.
Next, we perform survival analysis of the identified modules using the R-packages survival (Therneau and Lumley, 2017) and survcomp (Schröder et al., 2011). A multivariate Cox model is fitted with the sponge lncRNAs and mRNAs of the identified modules, and the fitted Cox model is used to predict the risk score of a sample. All the samples are divided into the high risk and the low risk groups equally according to their risk scores. We calculate Hazard Ratio (HR) between the high and the low risk groups, and the Log-rank test as well as the Kaplan Meier curve is also generated.
To further understand the underlying biological processes and pathways associated with the modules, the R-package clusterProfiler (Yu et al., 2012) is used to conduct functional enrichment analysis. The Gene Ontology (GO) (Ashburner et al., 2000) biological processes and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) pathways with adjusted P-value < 0.05 (adjusted by BH method) are considered as functional categories for the modules. In addition, we collect a list of lncRNAs and mRNAs associated with all four human cancers (GBM, LSCC, OvCa and PrCa) to investigate enriched cancer genes in each module. The list of lncRNAs related to the four human cancers is obtained from LncRNADisease v2015 (Chen et al., 2013), Lnc2Cancer v2016 (Ning et al., 2016) and MNDR v2013 (Wang et al., 2013). The list of mRNAs associated with the four human cancers is from DisGeNET v4.0 (Piñero et al., 2017), which is a comprehensive database integrating information on human disease-associated genes from several public databases and literatures.
3 Results
3.1 The LncmiRSRNs in human cancers
By following the steps of the LncmiRSRN method, we have constructed the sponge lncRNA-mRNA regulatory networks in four types of human cancers: GBM, LSCC, OvCa and PrCa (see Supplementary Material S2 for details), respectively. The numbers of sponge lncRNA-mRNA regulatory relationships in the four LncmiRSRNs are considerably different. However, the node degree distributions of the LncmiRSRNs in GBM, LSCC, OvCa and PrCa all fit power law distribution well with R2 > 0.95 (see Fig. 2A). This result indicates that the four LncmiRSRNs are scale free, similar to large-scale true biological networks.
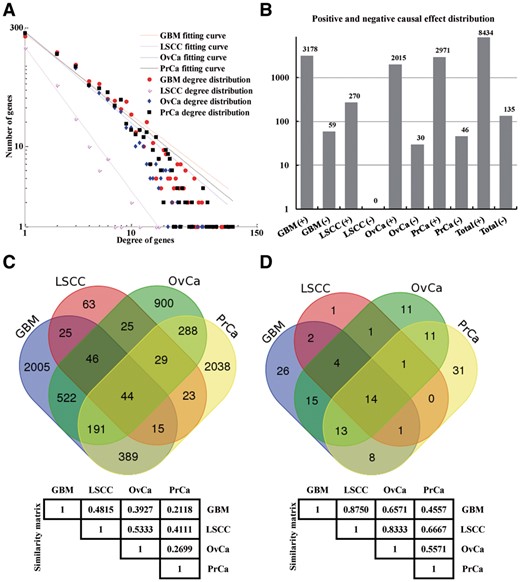
The four LncmiRSRNs identified for GBM, LSCC, OvCa and PrCa. (A) Degree distributions of the four LncmiRSRNs. (B) Causal effect distributions displaying that most sponge lncRNAs have positive effects on mRNAs. (C) Overlap and difference of sponge lncRNA-mRNA regulatory relationships in the four LncmiRSRNs and similarity matrix in terms of sponge lncRNA-mRNA regulatory relationships. (D) Overlap and difference of hub lncRNAs in the four LncmiRSRNs and similarity matrix in terms of hub lncRNAs
To uncover the role of sponge lncRNAs on mRNAs, we explore the causal effects which sponge lncRNAs have on mRNAs. As shown in Figure 2B, the number of positive sponge lncRNA-mRNA regulatory pairs is much larger than the negative sponge lncRNA-mRNA regualtory pairs in the four human cancers, implying that most sponge lncRNAs have positive effects on the expression levels of mRNAs. Moreover, it may indicate that through lncRNA-based miRNA sponging, mRNAs are normally up-regulated by sponge lncRNAs in LncmiRSRN.
As shown in Figure 2C and D, only a minority of sponge lncRNA-mRNA regulatory relationships (44, ∼0.67%) and hub lncRNAs (14, ∼10.07%) are shared by the four cancer LncmiRSRNs. This result suggests that a small portion of sponge lncRNA-mRNA regulatory relationships and hub lncRNAs tend to act as common miRNA sponge regulatory relationships and miRNA sponges that are involved in the biological processes of GBM, LSCC, OvCa and PrCa. On the other hand, a large number of sponge lncRNA-mRNA regulatory relationships (5006, ∼75.81%) and hub lncRNAs (69, ∼49.64%) are cancer-specific, indicating that many sponge lncRNA-mRNA regulatory relationships and hub lncRNAs selectively play a role in a specific human cancer.
In terms of sponge lncRNA-mRNA regulatory relationships and hub lncRNAs, we also calculate the similarity scores [Equation (1)] between each pair of the LncmiRSRNs in GBM, LSCC, OvCa and PrCa. In terms of sponge lncRNA-mRNA regulatory relationships, it is found that the similarity between the LncmiRSRN in LSCC and the LncmiRSRN in OvCa has the highest values (Sim = 0.5333). In terms of hub lncRNAs, the similarity between the LncmiRSRN in GBM and the LncmiRSRN in LSCC has the highest values (Sim = 0.8750). In particular, the findings in terms of hub lncRNAs suggest that GBM and LSCC may share similar hub lncRNAs for gene regulation. Furthermore, the similarity score in terms of sponge lncRNA-mRNA regulatory relationships is positively correlated with the similarity score in terms of hub lncRNAs (cor = 0.9594, P-value = 0.002). This result implies that the order of similarity between pairs of the LncmiRSRNs in GBM, LSCC, OvCa and PrCa using the two terms are normally consistent.
3.2 Network analysis reveals rewired and pivotal LncmiRSRNs across human cancers
Although the four cancer LncmiRSRNs share several common features, most sponge lncRNA-mRNA regulatory relationships in the LncmiRSRNs show a rewired mode between human cancers, like ‘on/off’ switches. From the above results, we observe that ∼75.81% sponge lncRNA-mRNA regulatory relationships only exist in one individual cancer, and only ∼0.67% sponge lncRNA-mRNA regulatory relationships are conserved in all four human cancers. The low conservation may be explained as that sponge lncRNA-mRNA regulatory relationships are more likely to be cancer-specific at expression level.
We merge differential and conserved sponge lncRNA-mRNA regulatory relationships to construct differential and conserved LncmiRSRNs (details in Supplementary Material S3). In Figure 3A, the node degree distributions of differential and conserved LncmiRSRNs fit power law distribution well with R2 = 0.9542 and 0.9914, respectively. This result suggests that in the differential and conserved LncmiRSRNs, most sponge lncRNAs have few interacting mRNAs while a small portion of sponge lncRNAs have many interacting mRNAs, and this characteristic is similar to that of most types of true biological networks.
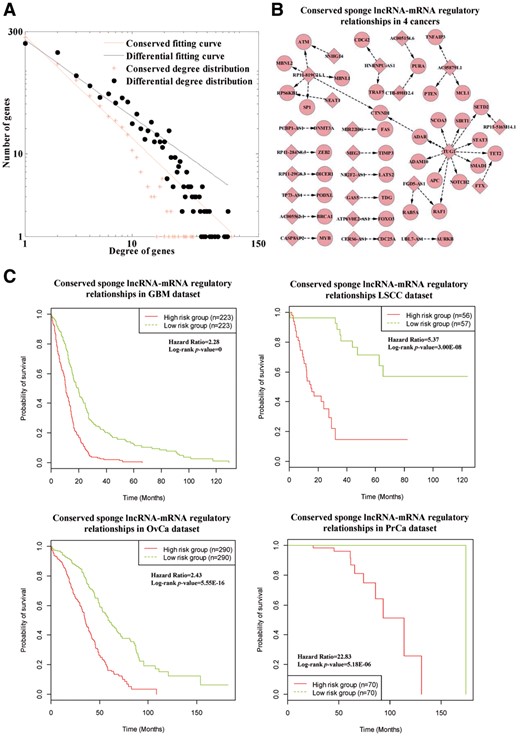
Differential and conserved LncmiRSRNs across human cancers. (A) The node degree distributions of differential and conserved LncmiRSRNs. (B) The pivotal sponge lncRNA-mRNA regulatory relationships existing in four human cancers. The red diamond and circle nodes denote sponge lncRNAs and mRNAs, respectively. (C) Survival analysis of conserved sponge lncRNA-mRNA regulatory relationships in GBM, LSCC, PrCa and OvCa datasets (Color version of this figure is available at Bioinformatics online.)
To evaluate whether there is a common pivot of sponge lncRNA-mRNA regulatory relationships to maintain the architecture of LncmiRSRNs across human cancers, we focus on investigating conserved sponge lncRNA-mRNA regulatory relationships in four human cancers (see Fig. 3B). Based on existing gene-disease associations (see Section 2.7), 5 lncRNAs and 35 mRNAs in the conserved LncmiRSRN are closely associated with at least one of the four cancers (see Table S1 in Supplementary Material S1 for details). Specifically, 14 mRNAs (APC, ATM, AURKB, CDC42, DNMT3A, FAS, MCL1, PTEN, RAF1, RPS6KB1, SIRT1, STAT3, SP1 and TIMP3) are involved in all four human cancers. In GBM dataset, the hazard ratio between the high and low risk groups based on the lncRNAs and mRNAs in the conserved LncmiRSRN is 2.28, and the Log-rank P-value is 0 (Fig. 3C). This result shows that the lncRNAs and mRNAs in the conserved LncmiRSRN can act as prognostic genes to discriminate the metastasis risks of GBM patients significantly. Moreover, the performance of the lncRNAs and mRNAs in the conserved LncmiRSRNs are also good in LSCC, OvCa and PrCa datasets (Fig. 3C). The hazard ratio is 5.37 and the Log-rank P-value is 3.00E-08 in LSCC dataset. As for OvCa dataset, the hazard ratio is 2.43 and the Log-rank P-value is 5.55E-16. In the PrCa dataset, hazard ratio is 22.83 and the Log-rank P-value is 5.18E-06. These findings indicate that the conserved LncmiRSRN may act as a common pivot to maintain the architecture of LncmiRSRNs across human cancers.
3.3 Differential and conserved hub lncRNAs are potential cancer drivers
According to the topological analysis of the LncmiRSRNs, a main characteristic of the four cancer LncmiRSRNs is that nodes in each LncmiRSRN have very different levels of degrees. Since it is found that hub nodes play important roles in biological networks, we identify hub lncRNAs that may act as potential cancer drivers in each LncmiRSRN. To systematically evaluate which hub lncRNAs are shared across different cancer-specific LncmiRSRNs, we divide hub lncRNAs into two categories (see ‘Topological properties of the LncmiRSRN’ section): differential and conserved hub lncRNAs. As a result, we have identified 69 differential and 70 conserved hub lncRNAs across cancers. Based on our collected gene-disease association databases (see Section 2.7), two out of 69 differential hub lncRNAs and nine out of 70 conserved hub lncRNAs are related to at least one of the four cancers (see Fig. 4A). In addition, most of the cancer-related hub lncRNAs (two out of the two, and six out of the nine in cancer-related differential and conserved hub lncRNAs, respectively) are related to at least two cancers. The result indicates that these differential and conserved hub lncRNAs as potential cancer drivers may influence the cancerous state in different human cancers.
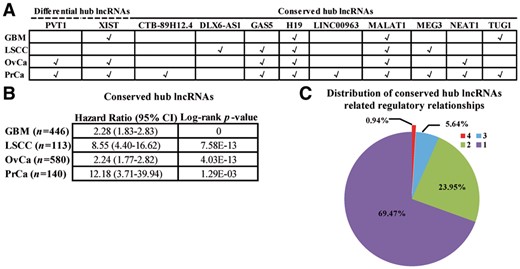
Differential and conserved hub lncRNAs between cancers. (A) Cancer-associated differential and conserved hub lncRNAs. (B) Multivariate survival analysis of conserved hub lncRNAs for the four human cancers. The forest plot shows hazard ratio (95% confidence interval), and the Log-rank P-values are <0.001. (C) Distribution of regulatory relationships associated with conserved hub lncRNAs across cancers
To explore whether there is a common core of hub lncRNAs to influence the cancerous state in different human cancers, we concentrate on conserved hub lncRNAs across human cancers. As shown in Figure 4B, survival analysis reveals that these conserved hub lncRNAs are significantly discriminative in dividing the metastasis risks of the four human cancers (Hazard Ratio > 2, Log-rank P-value < 0.01). Moreover, most conserved hub lncRNAs related regulatory relationships (∼69.47%) selectively tend to be cancer-specific (see Fig. 4C), suggesting that conserved hub lncRNAs may be involved in the biological processes of different human cancers through regulating different sets of targets. These findings imply that the conserved hub lncRNAs may act as a common core of potential cancer drivers to influence the cancerous state in different human cancers.
3.4 Functional annotation of differential and conserved LncmiRSRN network modules
Differential and conserved LncmiRSRNs display rewired and pivotal mode across human cancers, respectively. In the process of rewired and pivotal mode, network modules in differential and conserved LncmiRSRNs may work as functional units underlying complex human cancers. Thus, we are interested in identifying differential and conserved LncmiRSRN network modules. Network modules in differential and conserved LncmiRSRNs represent communities of functionally associated genes involved in specific biological processes. Investigating differential and conserved LncmiRSRNs can reveal several differential and conserved network modules of particular interest.
In total, the numbers of the identified differential and conserved LncmiRSRN network modules are 55 and 29, respectively (details in Supplementary Material S4). Functional enrichment analysis reveals that 52 out of the 55 (∼94.55%) differential modules and 28 out of the 29 (∼96.55%) conserved modules are significantly enriched in at least one GO biological process or KEGG pathway, respectively (see Table S2 in Supplementary Material S1 for details). This analysis suggests that most differential and conserved LncmiRSRN network modules work as functional units in at least one biological process. By mapping cancer-related genes to components of the differential and conserved LncmiRSRN network modules, we further investigate cancer gene enrichment in each module. As a result, all differential modules and conserved modules contain genes associated with at least one cancer, respectively (see Table S3 in Supplementary Material S1 for details). The result shows that all differential and conserved LncmiRSRN network modules may act as cancer-related modules.
3.5 Differential and conserved LncmiRSRN network modules can distinguish metastasis risks of human cancers
As illustrated above, most differential and conserved LncmiRSRN network modules work as functional units in at least one biological process and may act as cancer-related modules. Thus, these modules may be good module biomarkers. To demonstrate this assumption, we use the genes of each differential and conserved LncmiRSRN network module to predict the metastasis risks for GBM, LSCC, OvCa and PrCa patients. In this work, the modules with Hazard Ratio no <1.5 and Log-rank P-value < 0.05 are regarded as module biomarkers.
We find that the numbers of module biomarkers for the 55 differential modules in GBM, LSCC, OvCa and PrCa datasets are 14, 28, 13 and 15, respectively. In addition, in the 29 identified conserved modules, 3, 14, 2 and 10 conserved modules act as module biomarkers of GBM, LSCC, OvCa and PrCa, respectively (see Tables S4–S7 in Supplementary Material S1 for details). In total, the numbers of differential and conserved module biomarkers unique to only one cancer are 33 and 20, respectively. These findings suggest that differential and conserved LncmiRSRN network modules can act as module biomarkers to distinguish metastasis risks of cancers.
4 Discussions and conclusions
Growing evidence has shown that lncRNAs are emerging as key regulators of many biological processes in physiological and pathological states. Investigating the roles of lncRNAs acting as miRNA sponges provides a novel way to predict lncRNA functions. Based on ceRNA hypothesis, previous studies focus on studying indirect competing relationships between sponge lncRNAs and mRNAs. However, whether sponge lncRNAs can further regulate target mRNAs is still not unearthed.
In this work, we present a novel method to reveal the regulatory mechanism of sponge lncRNAs by considering the causal semantics of sponge lncRNA-mRNA relationships. For improving the calculating efficiency, we use the parallelized version of IDA to estimate the causal effects that a sponge lncRNA has on an mRNA.
We have applied our method to the four human cancer datasets: GBM, LSCC, OvCa and PrCa. To obtain a reliable candidate lncRNA-mRNA pairs, we collect several well-known experimentally miRNA-target interaction databases as ground-truth. The enrichment and survival analysis results show that the proposed method can help to elucidate sponge lncRNA related casual regulatory mechanisms of human cancers.
It is possible that lncRNAs are expressed at low levels, and it is also possible that the existing techniques may not be able to accurately measure lncRNA expression. However, since there have been known regulatory relationships between lncRNAs and mRNAs, such as those in the LncRNADisease database (Chen et al., 2013), we hypothesize that lncRNAs may regulate these mRNAs after the mRNAs being released by miRNAs. In other words, lncRNAs and miRNAs co-regulate the mRNAs (possibly at different timeframe). In our sponge model, the miRNAs (down) regulate the mRNAs to a certain level and release them. lncRNAs will then regulate the released mRNAs. This is different from the work looking at direct lncRNA-mRNA regulation. We hope our method and findings based on the above mentioned assumptions can provide high-confidence candidates of lncRNA-mRNA interactions for follow-up wet-lab experiments, thus contributing to the efforts on uncovering the mechanism of lncRNA-mRNA regulation.
There is still room to extend or improve our method. Firstly, LncmiRSRN is focused on lncRNA related miRNA sponge regulatory network in human cancer. To study all forms of aberrant lncRNA regulation, it is necessary to infer lncRNA related miRNA sponge regulatory network in human cancer and normal condition, respectively. Moreover, LncmiRSRN is sensitive to the number of samples, and has poor reproducibility in smaller subsets of the samples. It is noted that it is a common challenge of computational methods including our method. We plan to tackle this problem by using random sampling strategy, and separately identifying LncmiRSRN network in each subset of the samples. To obtain a robust LncmiRSRN, we will remove the sponge lncRNA-mRNA regulatory relationships that only exist k (e.g. 1) times in n (e.g. 10) different LncmiRSRNs. Additionally for each pair of putative lncRNA-mRNA, LncmiRSRN applies two commonly used principles (significant sharing of miRNAs at sequence level and positively correlated at expression level) to identify lncRNA related miRNA sponge interactions. However, some other factors, such as miRNA response elements (MREs) lying in RNA transcripts and miRNA regulation at expression level, may be also associated with the identification of lncRNA related miRNA sponge interactions. Our method is designed for the scenario in which miRNA expression data is lacking, but when miRNA expression data is available, its Step (1) can be replaced by an existing method which makes use of miRNA expression data in the identification of lncRNA related miRNA sponge networks. Finally, some of lncRNA related miRNA sponge interactions may be caused by transcriptional co-regulation (e.g. same promotors or transcription factors). This is a common problem with existing computational methods, including ours, and this problem may result in false positives. However, as the focus of this paper is on lncRNAs’ effect on the expression of the mRNAs after they are released from miRNA-mRNA interactions, we do not consider other possible factors in our study. We hope that the proposed method is still useful in shortlisting statistically significant interactions for follow-up wet lab experiments.
In summary, we propose a causality-based method to identify lncRNA related miRNA sponge regulatory network by integrating expression data, clinical information and miRNA-target interactions. To our best knowledge, this is the first method to study how the expression levels of the released mRNAs activate. Our method not only complements the ceRNA hypothesis, but provides a new avenue to study the functions and regulatory mechanism of lncRNAs in human cancers.
Funding
This work has been supported by the National Natural Science Foundation of China (61702069), the Applied Basic Research Foundation of Science and Technology of Yunnan Province (2017FB099), the NHMRC Grant (1123042) and the Australian Research Council Discovery Grant (DP170101306).
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}