





Abstract
Ion Torrent sequencing is one of the most frequently used platforms in healthcare research and industry. Despite many advantages, platform-specific artifacts complicate efficient separation of true variants from errors, especially in variants with lower allele frequencies (<15%). Here, we developed a multi-step filtering toolbox AIRVF that works on flowgram, raw and mapped reads and called variants to reduce artifact-driven false variant calls. Tests on sequencing data of standard reference material showed up to ∼98% reduction of false variants when combined to conventional public pipelines and ∼48% to the in-house commercial solution, with a minimal loss of sensitivity.
The program with a detailed manual is available at https://sourceforge.net/projects/airvf/.
Supplementary data are available at Bioinformatics online.
1 Introduction
Ion semiconductor sequencing, such as Ion Torrent PGM is one of the primary options in a broad area of genomic research and clinic in light of the lower instrument cost, short operation time and smaller sample requirement. Besides, higher error rates, premature sequence truncation and inaccurate determination of homopolymer repeats are known drawbacks that are genuine to the sequencing technology (Salipante et al., 2014). As a result, the reported accuracy in calling single nucleotide variations (SNV) and indels in Ion Torrent platform is lower than in Illumina platform (Hwang et al., 2015).
So far, a few studies directly interrogated the problem to improve pipelines for detection of germ line SNVs and indels in Ion Torrent sequencing (Zeng et al., 2013; Zhu et al., 2014). Such continuous efforts secured a robust performance in a routine analysis of germ line variant in a focused gene set (Zanella et al., 2017). However, there is still a lack of evaluation and optimization of pipelines for somatic mutations, whose signatures are frequently compromised in data due to heterogeneity or contamination issues in a sample, thereby lowering variant allele frequencies (VAF) down to ∼10% (Cibulskis et al., 2013). As variants with lower VAFs are likely undetectable by germ line detection algorithms (target VAF = 50%), utilization of well-proven, somatic mutation callers is desired for accurate variant analysis (Koboldt et al., 2012).
However, a naïve application of conventional methods to a heterogeneous platform is discouraged due to a potential large drop in accuracy. Currently, the vendor’s in-house solution, the Torrent Suite Software (TSS), is considered the only specialized program for somatic variant calling through its components including TMAP (aligner), Torrent Variant Caller (TVC, variant caller) and Ion Reporter (IR, variant filter/analyzer). However, the detailed algorithm with a strictly measured performance has not been reported publicly, and the tight binding of the software to its commercial server also complicates a general use.
AIRVF is an integrated, multi-step filtering toolbox that can be applied prior to conventional and commercial somatic variant calling pipelines to secure compatibility and improve accuracy by removing platform-specific artifacts from the data. We expect that the use of AIRVF would not only reduce false variants but also increase the utility of the platform by expanding the choice of analysis tools.
2 Methods and implementation
AIRVF implements a set of Ion Torrent sequencing filters that work on three different levels: (i) raw-reads, (ii) mapped-reads and (iii) called variants. Raw-read filters access the raw sequencing data (FASTQ) to remove or trim unreliable reads and bases. To secure the minimum throughput for variant calling, filters were designed to retain at least 50% of the total data (Supplementary Fig. S1). Mapped-read filters interrogate alignment files (BAM) to presume erroneous regions, based on the observed positive association between false positive rates and the number of indels in mapped reads (Supplementary Fig. S2). Called variant filters take a list of variants (VCF) to identify false calls based on multiple erroneous signatures including a chi-square based direction-specific errors (Shin and Park, 2016) (Supplementary Table S1). The types of filters and their target errors are described below:
Raw-read filters
· contaminant filter Identifies and removes sample contaminants.
· flowgram filter Removes reads with ambiguous flow values (which determines the length of homopolymer).
· read length filter Removes abnormally short reads resulted from a possible DNA fragment extension error.
· quality filter Removes bases with low base-call quality. Average and minimum thresholds are applied.
· quality trimming Trims reads with possibly damaged primers.
Mapped-read filters
· per-read indel filter Removes reads with indels.
Variant filters
· allele-based filter Filters out variants based on multiple allele-related values including read depth, allele frequency and count and strand bias.
· sample filter Filters out formalin-fixed and paraffin-embedded (FFPE) sample specific error signatures.
To determine the optimized values for the filtering criteria, we used two sequencing data resources for the implementation of the filters. First, a publicly available microbial data (non-heterozygous) set was downloaded and assessed to determine base-call quality filters. Second, we directly sequenced a standard reference material (Acrometrix Oncology Hotspot Control, AOHC) that contains 555 engineered true variants (SNVs and indels) with a low VAF (5–15%). Based on the data, distributions of erroneous reads and bases, ambiguity in flowgram values, positional quality scores, mapping quality and the effect of homopolymer in false variants were analyzed and used to determine optimal parameters for filtration (see Supplementary Methods M1 to M4 for more details). Finally, an independent additional sequencing of AOHC was performed for validation of AIRVF.
3 Results and discussion
The effects of AIRVF in the somatic variant analysis are shown in Figure 1. We applied different levels of filtering on the training data (Fig. 1A). Application of raw-read and mapped-read level filters gradually reduced false calls (21–25k and 15–18k/Mbp, respectively) from no filtered data (∼40k/Mbp) with a minimal loss of sensitivity (0.2 and 0.4%, respectively), regardless of the alignment programs. The variant-level filters remarkably removed false calls (534–801, 97.7–98.6% reduction of FPR) with a slight loss of sensitivity (4.2–5.4%). The user-configurable filters enable a flexible application of AIRVF regarding the type of analysis.
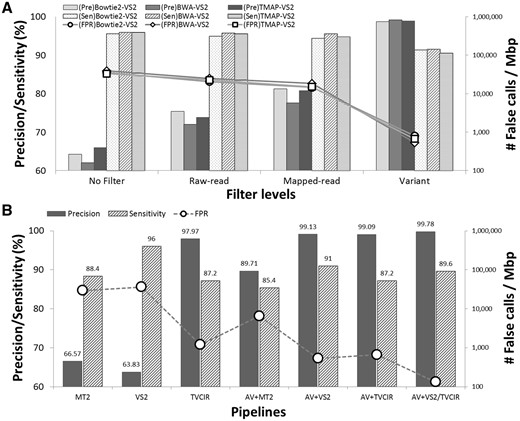
Effects of AIRVF in somatic variant calling. (A) Precision (Pre), sensitivity (Sen) and false positive rate (FPR) are measured in AOHC standard reference. VarScan2 (VS2) was used with three different aligners (Bowtie2, BWA-MEM and TMAP). Different levels of filters were cumulatively applied to observe the effect. When entirely applied, a remarkable increase of precision was acquired (solid bars), with a slight loss of sensitivity (patterned bars). The results correspond to a ∼99% reduction of false calls (marked lines, right axis in logarithmic scale). (B) Performance of pipelines in an independent AOHC sequencing. Compared to the simple use of somatic callers: MuTect2 (MT2) and VarScan2 (VS2), applying AIRVF (AV+) showed a compatible or superior performance to the commercial in-house pipeline (TVCIR), which can be further refined by AIRVF without a single loss of true calls (AV + TVCIR). Use of multiple callers almost eliminated false calls (AV + VS2/TVCIR) almost eliminated platform-driven false calls. BWA-MEM was used as a default mapping tool
Next, we applied AIRVF with general somatic variant callers, MuTect2 (MT2) and VarScan2 (VS2), and the vendor’s software TVC with IR filtration (TVCIR) (Fig. 1B); the single use of TVC led to a worse precision (33.5%) with a similar sensitivity (89.2%). As expected, the direct use of the conventional program on Ion Torrent sequencing resulted in a low precision (66.6% in MT2, 63.8% in VS2), which correspond to an FPR of 30k and 36k/Mbp. By applying AIRVF, the precision was increased to 89.7% (MT2), and 99.1% (VS2) with a slight loss of sensitivity (1.2% in MT2, and 5% in VS2). These accuracy values were compatible or higher to the TVC-IR pipeline, confirming that AIRVF successfully reduced platform-specific artifacts for the use of general variant callers. In addition, AIRVF further increased the precision of TVF-IR from 97.97 to 99.09% (corresponds to a 48% reduction of false calls) without a single loss of true variant, respectively. Based on the differed performance of TVCIR and VS2 in calling SNVs and indels, the combination of two tools (VS2 for SNV and TVCIR for indel) with AIRVF almost eliminated false positive calls (133/Mbp) with maintaining sensitivity. The complete analysis results are described in the Supplementary Material (Supplementary Figs S1 to S15 and Tables S1–S10).
Development of AIRVF based on the standard reference confirmed the risk of overwhelming false variant calls in conventional pipelines, which can be efficiently reduced with proper filtration of sequencing data. Also, we expect that utilization of publicly available tools would expedite the methodological advances that lead to a better application of the platform.
Acknowledgement
We thank Dr. Junsu Ko (Future Medicine Co., Ltd.) for technical advice.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning [2015R1C1A1A01053638] and the Health Technology R&D Project through the Korea Health Industry Development Institute funded by Ministry of Welfare [HI14C1324 and HI15C1601]. SK was funded by the research grant from the Yonsei University College of Medicine [6-2016-0081].
Conflict of Interest: none declared.
References


{kind=link}