





Abstract
Predicting the secondary structure of an ribonucleic acid (RNA) sequence is useful in many applications. Existing algorithms [based on dynamic programming] suffer from a major limitation: their runtimes scale cubically with the RNA length, and this slowness limits their use in genome-wide applications.
We present a novel alternative O(n3)-time dynamic programming algorithm for RNA folding that is amenable to heuristics that make it run in O(n) time and O(n) space, while producing a high-quality approximation to the optimal solution. Inspired by incremental parsing for context-free grammars in computational linguistics, our alternative dynamic programming algorithm scans the sequence in a left-to-right (5′-to-3′) direction rather than in a bottom-up fashion, which allows us to employ the effective beam pruning heuristic. Our work, though inexact, is the first RNA folding algorithm to achieve linear runtime (and linear space) without imposing constraints on the output structure. Surprisingly, our approximate search results in even higher overall accuracy on a diverse database of sequences with known structures. More interestingly, it leads to significantly more accurate predictions on the longest sequence families in that database (16S and 23S Ribosomal RNAs), as well as improved accuracies for long-range base pairs (500+ nucleotides apart), both of which are well known to be challenging for the current models.
Our source code is available at https://github.com/LinearFold/LinearFold, and our webserver is at http://linearfold.org (sequence limit: 100 000nt).
Supplementary data are available at Bioinformatics online.
1 Introduction
Ribonucleic acid (RNA) is involved in numerous cellular processes (Eddy, 2001). The dual nature of RNA as both a genetic material and functional molecule led to the RNA World hypothesis, that RNA was the first molecule of life (Gilbert, 1986), and this dual nature has also been utilized to develop in vitro methods to evolve functional sequences (Joyce, 1994). Furthermore, RNA is an important drug target and agent (Angelbello et al., 2018; Castanotto and Rossi, 2009; Childs-Disney et al., 2007; Crooke, 2004; Gareiss et al., 2008; Palde et al., 2010; Sazani et al., 2002).
Predicting the secondary structure of an RNA sequence, defined as the set of all canonical base pairs (A–U, G–C, G–U, see Fig. 1A), is an important and challenging problem (Hofacker and Lorenz, 2014; Seetin and Mathews, 2012). Knowing the structure reveals crucial information about the RNA’s function, which is useful in many applications ranging from ncRNA detection (Fu et al., 2015; Gruber et al., 2010; Washietl et al., 2012) to the design of oligonucleotides for knockdown of message (Lu and Mathews, 2008; Tafer et al., 2008). Since experimentally determining the structure is expensive and time-consuming, and given the overwhelming increase in genomic data (about 1021 base-pairs per year) (Stephens et al., 2015), computational methods have been widely used as an alternative to automatically predict the structure. Widely used systems such as RNAstructure (Mathews and Turner, 2006), Vienna RNAfold (Lorenz et al., 2011), CONTRAfold (Do et al., 2006) and CentroidFold (Sato et al., 2009), all use virtually the same dynamic programming algorithm (Nussinov et al., 1978; Zuker and Stiegler, 1981) to find the best-scoring (lowest free energy, maximum expected accuracy or best model score) structure (Mathews and Turner, 2006; Washietl et al., 2012). However, this set of algorithms, borrowed from computational linguistics (Kasami, 1965; Younger, 1967), has a running time of O(n3) that scales cubically with the sequence length n, which is too slow for long RNA sequences (Lange et al., 2012).
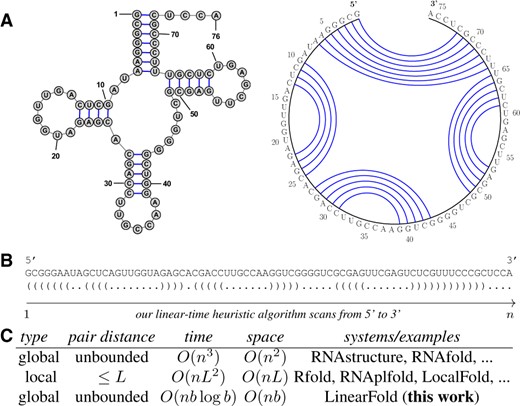
Summary of our work. (A) Secondary structure representations of E.coli tRNAGly; (B) the corresponding dot-bracket format and an illustration of our algorithm, which scans the sequence left-to-right, and tags each nucleotide as ‘.’ (unpaired), ‘(’ (to be paired with a future nucleotide) or ‘)’ (paired with a previous nucleotide). (C) Comparison between our work and existing ones. L is the limit of pair distance in local folding methods (often ≤150), and b is the beam size in our work (default 100). Our algorithm, though approximate, is the first to achieve linear runtime without imposing constraints on the output structure
As an alternative, faster algorithms that predict only a restricted subset of structures have been proposed. On the one hand, local folding methods such as Rfold (Kiryu et al., 2008), Vienna RNAplfold (Bernhart et al., 2006) and LocalFold (Lange et al., 2012) run in linear time but only predict base pairs up to L nucleotides apart ( in the literature; see Fig. 1C). On the other hand, due to the prohibitive cubic runtime of standard methods, it has been a common practice to divide long RNA sequences into short segments (e.g. ≤700nt) and predict structures within each segment only (Andronescu et al., 2007; Licon et al., 2010; Watts et al., 2009). All these local methods omit long-range base pairs, which theoretical and experimental studies have demonstrated to be common in natural RNAs, especially between the 5′ and 3′ ends (Lai et al., 2018; Li and Reidys, 2018; Seetin and Mathews, 2012).
We instead design LinearFold, an approximate algorithm that is the first in RNA folding to achieve linear runtime (and linear space) without imposing constraints on the output structure such as base pair distance. While the classical O(n3)-time algorithm is bottom-up, making it hard to linearize, ours runs left-to-right (i.e. 5′-to-3′), incrementally tagging each nucleotide in the dot-bracket format (Fig. 1B). While this naive version runs in the exponential time of O(3n), we borrow an efficient packing idea from computational linguistic (Tomita, 1988) that reduces the runtime back to O(n3). This novel left-to-right O(n3) dynamic program is also a contribution of this article. Furthermore, on top of this exact algorithm, we apply beam search, a popular heuristic to prune the search space (Huang and Sagae, 2010), which keeps only the top b highest-scoring (or lowest energy) states for each prefix of the input sequence, resulting in an time approximate search algorithm, where b is the beam size chosen by the user.
Our approach can ‘linearize’ any dynamic programming-based pseudoknot-free RNA folding system. In particular, we demonstrate two versions of LinearFold, LinearFold-V using the thermodynamic free energy model (Mathews et al., 2004) from Vienna RNAfold (Lorenz et al., 2011), and LinearFold-C using the machine learned model from CONTRAfold (Do et al., 2006). We evaluate our systems on a diverse dataset of RNA sequences with well-established structures, and show that while being substantially more efficient, LinearFold leads to even higher average accuracies over all families, and more interestingly, LinearFold is significantly more accurate than the exact search methods on the longest families, 16S and 23S Ribosomal RNAs. In addition, LinearFold is also more accurate on long-range base pairs, which is well known to be a challenging problem for the current models (Amman et al., 2013).
Finally, our work establishes a new connection among computational linguistics, compiler theory and RNA folding (see Supplementary Fig. SI 7).
2 The LinearFold algorithm
2.1 Problem formulation
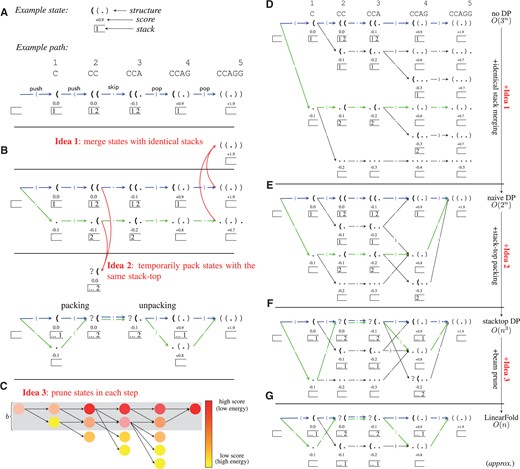
Illustration of the LinearFold approach, using a short sequence CCAGG and the simple scoring function (Eq. 2). (A) An example state and an example (actually optimal) path, showing states (predicted prefix structures), actions ( ‘(’, ‘.’, and ‘)’), and stacks (unpaired open brackets, which are shown in bold in states). (B) Two example paths (the optimal one in blue and a suboptimal one in green) and two essential ideas of left-to-right dynamic programming: merging equivalent states with identical stacks (Idea 1) and packing temporarily equivalent states sharing the same stack top, and corresponding unpacking upon (Idea 2). (C) Illustration of beam search, which keeps top b states (those in the shaded region) per step (Idea 3). (D) The whole search space of the naive algorithm (O(3n) time). (E) Improving to O(2n) time with Idea 1. (F) Further improving to O(n3) time with Idea 2. (G) Further improving to O(n) time (but with approximate search) with Idea 3. In B, F, and G, each green/blue arrow pair ?( ?(. is actually a single arrow, denoting two paths temporarily packed as one; we draw paired arrows to highlight that two states .( and (( are performing action together. Note the version up to Idea 2 is exact and worst-case O(n3) time
?(. is actually a single arrow, denoting two paths temporarily packed as one; we draw paired arrows to highlight that two states .( and (( are performing action together. Note the version up to Idea 2 is exact and worst-case O(n3) time
In reality, however, the actual scoring functions used by CONTRAfold, RNAfold, and our LinearFold are much more complex, and they decompose into individual loops. See Supplementary section B for details.
2.2 Idea 0: Brute-force search: O(3n)
- : label as ‘(’ for it to be paired with a downstream nucleotide, and pushing j + 1 on to the stack, notated:
- : label as ‘.’ (unpaired and skipped):
- : label as ‘)’, paired with the upstream nucleotide xi where i is the top of the stack, and pop i (if pair is allowed):
We start with the init state and finish with any state with an empty stack (ensuring the output is a well-balanced dot-bracket sequence). See Figure 2A for an example path for input sequence CCAGG, and Figure 2D for all valid paths.
The above procedure describes a naive exhaustive search without dynamic programming which has exponential runtime O(3n), as there are up to three actions per step (see Fig. 2D).
Next, Figure 2B sketches the two key dynamic programming ideas that speed up this algorithm to O(n3) by merging and packing states.
2.3 Idea 1: merge states with identical stacks: O(2n)
2.4 Idea 2: pack temporarily equivalent states: O(n3)
We further observe that even though some states have different stacks, they might share the same stack top. For example, in step 2, ‘.(’ and ‘((’ have [2] and [1, 2] as their stacks, resp., but with the same stack top 2. Our key insight is that two states with the same stack-top are ‘temporarily equivalent’ and can be ‘packed’ as they would behave equivalently until the stack-top open bracket is closed (i.e. matched), after which they ‘unpack’ and diverge. As shown in Fig. 2B (Idea 2), both ‘.(’ and ‘((’ are looking for a ‘G’ to match with the stack top x2=‘C’, and can be packed as ‘?(’ with stack […2] where ‘?’ and ‘…’ represent histories that are not important for now. After skipping the next nucleotide x3=‘A’, they become ‘?(.’ and upon matching the next nucleotide x4=‘G’ with the stack-top x2=‘C’, they unpack, resulting in ‘.(.)’ and ‘((.)’.
2.5 Idea 3 (approximate search): beam pruning: O(n)
We further employ beam pruning (Huang et al., 2012), a popular heuristic widely used in computational linguistics, to reduce the complexity from O(n3) to O(n), but with the cost of exact search. Basically, at each step j, we only keep the b top-scoring (lowest-energy) states and prune the other, less promising, ones (because they are less likely to be part of the optimal final structure). This results in an approximate search algorithm in O(nb2) time, depicted in Figure 2C and G. On top of beam search, we borrow k-best parsing (Huang and Chiang, 2005) to reduce the runtime to . Here, the beam size b is a small constant (by default 100) so the overall runtime is linear in n. We will show that our approximate search achieves even higher overall accuracy than the classical exact search methods. The space complexity is O(nb). See Supplementary Figure SI 6 for the real system. There are two minor restrictions in our real system: the length of an interior loop is bounded by 30nt (a standard limit found in most existing RNA folding software such as CONTRAfold), so is the leftmost (5′-end) unpaired segment of a multiloop (new constraint). These conditions are valid for 37°C, and no violations were found in the ArchiveII dataset.
3 Results
3.1 Efficiency and scalability
We compare LinearFold’s efficiency with classical cubic-time algorithms represented by CONTRAfold (Version 2.02) and Vienna RNAfold (Version 2.4.10) (http://contra.stanford.edu/ and https://www.tbi.univie.ac.at/RNA/download/sourcecode/2_4_x/ViennaRNA-2.4.10.tar.gz). We use two datasets: (i) the ArchiveII dataset (Sloma and Mathews, 2016), a diverse set of RNA sequences with known structures (http://rna.urmc.rochester.edu/pub/archiveII.tar.gz; we removed those sequences found in the S-Processed set, see Supplementary Table SI 1 for details), and (ii) a sampled subset of RNAcentral (The RNAcentral Consortium, 2017) (https://rnacentral.org/), a comprehensive set of ncRNA sequences from many databases. While ArchiveII contains sequences of 3000nt or less, RNAcentral has many much longer ones, with the longest being 244 296nt (Homo Sapiens Transcript NONHSAT168677.1, from the NONCODE database (Zhao et al., 2016)). We run all programs (compiled by GCC 4.9.0) on Linux, with 3.40GHz Intel Xeon E3-1231 CPU and 32G memory.
Figure 3A shows that on the relatively short ArchiveII set, LinearFold’s runtime scales almost linearly with the sequence length, while the two baselines have superquadratic runtimes. On the much longer RNAcentral set, Figure 3B shows strictly linear runtime for LinearFold and near-cubic runtimes for the baselines, which agrees with the asymptotic analyses and suggests that the minor deviations from the theoretical runtimes are due to the short sequence lengths in the ArchiveII set. For a sequence of ∼10 000nt (e.g. the HIV genome), LinearFold takes only 8 s, while the baselines take 4 min. For a sequence of 32 753nt, LinearFold takes 26 s, while CONTRAfold and RNAfold take 2 and 1.7 h, respectively.
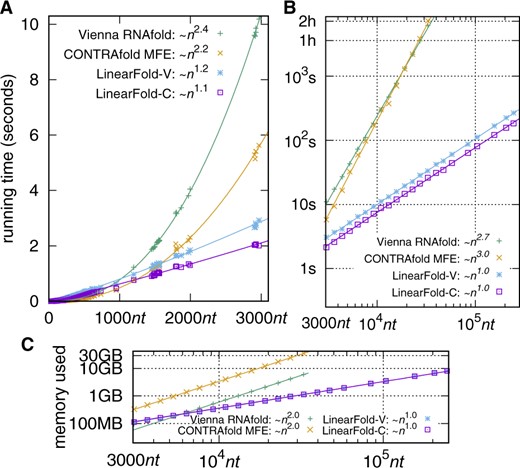
Efficiency and scalability of LinearFold. (A) Runtime comparisons on the ArchiveII dataset with the two baselines, CONTRAfold MFE and Vienna RNAfold. (B) Runtime comparisons on the RNAcentral dataset (log–log). (C) Memory usage comparisons (RNAcentral set, log–log). LinearFold uses O(n) time and memory, being substantially faster and slimmer than the O(n3)-time, O(n2)-space, baselines on long sequences
In addition, LinearFold uses only O(n) memory (Fig. 3C). The classical O(n3)-time algorithm uses O(n2) space, because it needs to solve the best-scoring substructure for each substring [i, j] bottom-up. LinearFold, in contrast, uses O(n) space thanks to left-to-right beam search, and is the first O(n)-space algorithm to be able to predict base pairs of unbounded distance. It is able to fold the longest sequence in RNAcentral (244 296nt) within 3 min, while neither CONTRAfold or RNAfold runs on anything longer than 32 767nt due to datastructure limitations. As a result, the sequence limit on our web server (105nt, see abstract) is 10× that of RNAfold web server (the previous largest), being by far the largest limit among all available servers (as of March 2019). The curve-fittings in Figure 3 were done log-log in gnuplot with in A, in B, and in C, to focus on the asymptotics.
3.2 Accuracy
We next compare LinearFold with the two baselines in accuracy, reporting both positive predictive value (PPV, the fraction of predicted pairs in the known structure) and sensitivity (the fraction of known pairs predicted) on each RNA family in the ArchiveII dataset, allowing correctly predicted pairs to be offset by one position for one nucleotide as compared to the known structure (Sloma and Mathews, 2016); we also report exact match accuracies in Supplementary Table SI 2. We test statistical significance using a paired, one-sided permutation test, following (Aghaeepour and Hoos, 2013).
Figure 4 shows that LinearFold is more accurate than the baselines, and interestingly, this advantage is more pronunced on longer sequences. Individually, LinearFold-C (the LinearFold implementation of the CONTRAfold model) is significantly more accurate in sensitivity than CONTRAfold on one family (Group I Intron), and both PPV/sensitivity on two families (16S and 23S ribosomal RNAs), with the last two being the longest families in this dataset, where they have average lengths 1548nt and 2927nt, and enjoyed +3.56%/+3.09% and +8.65%/+5.66% (absolute) improvements in PPV/sensitivity, respectively. LinearFold-V (the LinearFold implementation of the Vienna RNAfold model) also outperforms RNAfold with significant improvements in PPV on two families (SRP and 16S rRNA), and both PPV/sensitivity on one family (Group I Intron). Overall (across all families), LinearFold-C outperforms CONTRAfold by +1.3%/+0.9% PPV/sensitivity, while LinearFold-V outperforms RNAfold by +0.3%/+0.2%. See Supplementary Table SI 1 for details.
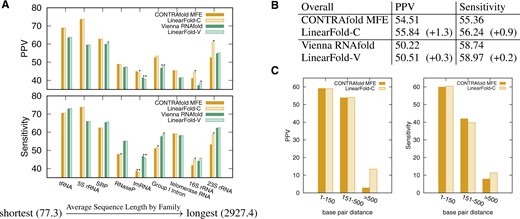
Accuracy of LinearFold. (A) Each bar represents PPV/sensitivity averaged over all sequences in one family. Statistical significance is marked as or . See Table 2 for details. (B) The overall accuracies, averaging over all families. (C) Each bar represents the overall PPV/sensitivity of all base pairs in a certain length range across all sequences. Supplementary Figure SI 1. shows a similar result for LinearFold-V. Overall, LinearFold outperforms exact search baselines, especially on longer families and long-range pairs
Long-range base pairs are notoriously difficult to predict under current models (Amman et al., 2013). Interestingly, LinearFold is more accurate in both PPV and sensitivity than the exact search algorithm for long-range base pairs of nucleotides greater than 500 nucleotides apart, as shown in Figure 4C. Combined with Supplementary Figure SI 1, we conclude that LinearFold is more selective in predicting long-range base pairs (higher PPV), but nevertheless predicts more such pairs that are correct (higher Sensitivity). Supplementary Figure SI 2B and C further shows that both LinearFold-C and LinearFold-V correct the severe overprediction of those long-range base pairs in exact search baselines.
Interestingly, even though our algorithm scans 5′-to-3′, the accuracy does not degrade toward the 3′-end, shown in Supplementary Figure SI 4.
3.3 Search quality
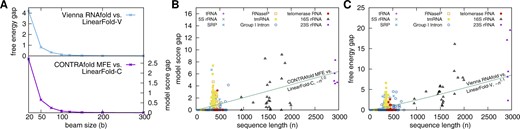
Search error (model score gap or free energy gap ΔΔG). (A) Average free energy gap (Vienna RNAfold vs. LinearFold-V) and model cost gap (CONTRAfold vs. LinearFold-C) with varying beam size; the search error shrinks with beam size, quickly converging to 0. (B and C) The search error (or gap) grows linearly with sequence length. Here, tmRNA is the outlier with disproportionally severe search errors, which can explain the slightly worse accuracies of LinearFold on tmRNA in Figure 4A. See Supplementary Figure SI 3. for a close-up on short sequences
3.4 Impacts of beam size on prediction accuracy
Figure 6A plots PPV and sensitivity as a function of beam size. LinearFold-C outperforms CONTRAfold MFE in both PPV and sensitivity with 75 and is stable with . Figure 6B shows the tradeoff between PPV and sensitivity. Both PPV and sensitivity increase initially with beam size, culminating at b = 120, and then decrease, converging to exact search. We do not tune the beam size on any dataset and use the round number of 100 as default. Figure 6C–D shows a similar trend for LinearFold-V.

Impacts of beam size on prediction accuracy. (A and C) PPV and sensitivity with varying beam size for LinearFold-C (A) and LinearFold-V (C); (B and D) PPV-sensitivity tradeoff for LinearFold-C (B) and LinearFold-V (D). Note that LinearFold with b = ∞ is exact search in O(n3) time (Idea 2) and produces identical results to the baselines
3.5 Example predictions: Group I intron, 16S & 23S rRNAs
Figure 7 visualizes the predicted secondary structures from three RNA families: Cryptothallus mirabilis Group I Intron, Bacillus subtilis 16S rRNA and Escherichia coli 23S rRNA. We observe that LinearFold substantially reduces false positives (shown in red), especially on the CONTRAfold model. It also correctly predicts many (clusters of) long-range base pairs (true positives, shown in blue), e.g. in C.mirabilis Group I Intron with LinearFold-C (Fig. 7D, pair distance 237nt), B.subtilis 16S rRNA with LinearFold-C (Fig. 7E, pair distance 460nt), E.coli 23S rRNA with both LinearFold-C and LinearFold-V (Fig. 7F and L, pair distance 582nt). This reconfirms LinearFold’s advantage in predicting long-range base pairs shown in Figure 4C. Moreover, LinearFold is able to predict the longest 5′–3′ pairs, as shown in E. coli 23S rRNA with LinearFold-V (Fig. 7L, pair distance 2901nt). In most cases (except LinearFold-V on B. subtilis 16S rRNA, Fig. 7K), LinearFold improves substantially over the corresponding baselines. By contrast, local folding methods do not predict any long-range pairs, shown in Fig. 8. We use RNAfold –maxBPspan 150 for local folding, and this limit of 150 is the largest default limit in the local folding literature and softwares.
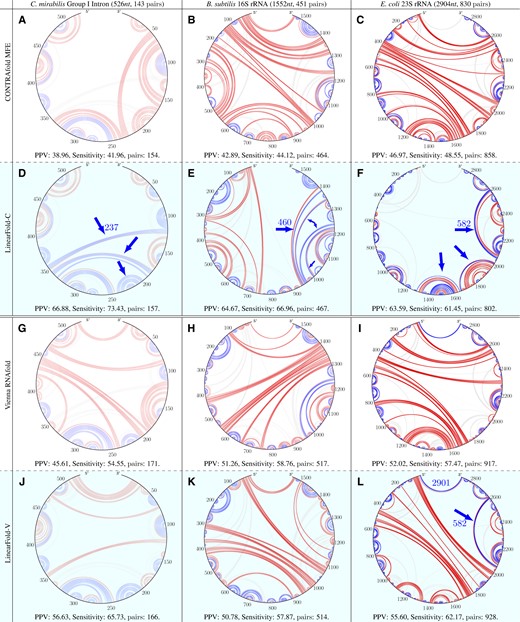
Circular plots of the prediction results on three RNA sequences (from three different RNA families) comparing the baselines (A–C: CONTRAfold MFE; G–I: Vienna RNAfold) and our LinearFold (D–F: LinearFold-C; J–L: LinearFold-V). Correctly predicted base pairs are in blue (true positives), incorrectly predicted pairs in red (false positives) and missing true base pairs in light gray (false negatives). Each plot is clockwise from 5′ to 3′. We can observe that (i) our LinearFold greatly reduces the false positives, especially on CONTRAfold; (ii) our LinearFold correctly predicts many long-range pairs, e.g. LinearFold-C on all three sequences and LinearFold-V on E.coli 23S rRNA(L); (iii) our LinearFold is able to predict the longest 5′–3′ pairs, even with the beam size of 100, which is an order of magnitude smaller than the sequence lengths of 16S and 23S rRNAs. (iv) In almost all cases (except for LinearFold-V on B.subtilis 16S rRNA(K)), LinearFold substantially outperforms the corresponding baseline
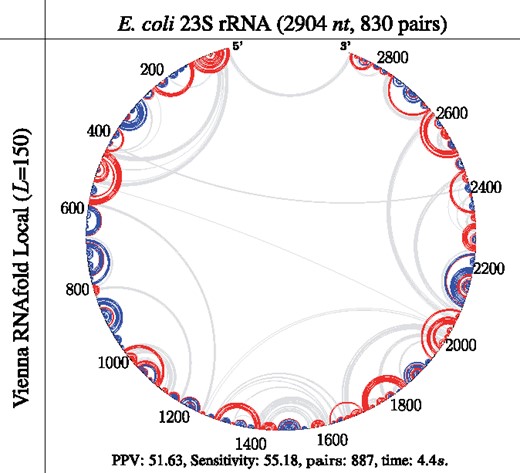
Circular plots of prediction results using the local folding mode of Vienna RNAfold (which only predicts local pairs no more than 150nt apart) on the E.coli 23S rRNA (corresponding to Fig. 7I). Moreover, the O(nL2)-time local folding (with default L = 150) is twice as slow as the -time LinearFold-V (with default b = 100)
4 Discussion
There are several reasons why our beam search algorithm, though approximate, outperforms the exact search baselines in terms of accuracy (esp. in 16S and 23S rRNAs and long-range base pairs).
First, the scoring functions are imperfect, so it is totally possible for a suboptimal structure (in terms of model score or free energy) to be more accurate than the optimal-score structure. For example, it was well-studied that while the lowest free energy structure contains only 72.9% of the actual base pairs (given a dataset), a structure containing 86.1% of them can be found with a free energy within 4.8% of the optimal structure (Mathews et al., 1999; Zuker et al., 1991).
Secondly, the beam search algorithm prunes lower-scoring (sub)structures at each step, requiring the surviving (sub)structures and the final result to be highly scored for each prefix. Our results suggest that this extra constraint, like ‘regularization’, could compensate for the inaccuracy of the (physical or machine-learning) model, as LinearFold systematically picks a more accurate suboptimal structure without knowing the ground truth; indeed, this seemingly surprising phenomenon has been observed before in computational linguistics (Huang and Sagae, 2010) which inspired this work.
Finally, our LinearFold algorithm resembles cotranscriptional folding where RNA molecules start to fold immediately before being fully transcribed (Gultyaev et al., 1995; Meyer and Miklós, 2004). This is analogous to psycholinguistic evidence that humans incrementally parse a sentence before it is fully read or heard (Frazier and Rayner, 1982). We hypothesize that some RNA sequences have evolved to fold co-transcriptionally (Meyer and Miklós, 2004), thus making our 5′-to-3′ incremental approach more accurate than bottom-up baselines. Supplementary Figure SI 5B shows a slight preference for 5′-to-3′ order over 3′-to-5′.
There are other algorithmic efforts to speed up RNA folding, including an algorithm using the Four-Russians method (Venkatachalam et al., 2014), and two other sub-cubic algorithms inspired by fast matrix multiplication and context-free parsing (Bringmann et al., 2016; Zakov et al., 2011). We note that all of them are based on the classical cubic-time bottom-up algorithm, and thus orthogonal to our left-to-right approach. There also exists a linear-time algorithm (Rastegari and Condon, 2005) to analyze a given structure, but not to predict one de novo.
5 Conclusion and future work
We designed an O(n)-time, O(n)-space, approximate search algorithm, using incremental dynamic programming plus beam search, and apply this algorithm to both machine-learned and thermodynamic models. Besides the linearity in both time and memory (Fig. 3), we also found:
Though LinearFold uses only a fraction of time and memory compared with existing algorithms, our predicted structures are even more accurate overall in both PPV and sensitivity and on both machine-learned and thermodynamic models (see Fig. 4).
The accuracy improvement of LinearFold is more pronunced on longer families such as 16S and 23S rRNAs (see Figs 4 and 7).
LinearFold is also more accurate than the baselines at predicting long-range base pairs over 500nt apart (Fig. 4C), which is well known to be challenging for the current models (Amman et al., 2013).
Although the performance of LinearFold depends on the beam size b, the number of base pairs and the accuracy of prediction are stable when b is in the range of 100–200.
There is a crucial difference between our LinearFold and local folding algorithms (Bernhart et al., 2006; Kiryu et al., 2008; Lange et al., 2012) that can only predict pairs up to a certain distance. Theoretical and empirical studies found several evidences that unboundedly long-distance pairs are actually quite common in natural RNA structures: (i) the length of the longest base pair grows nearly linearly with sequence length n (Li and Reidys, 2018); (ii) the physical distance between the 5′–3′ ends in folded structures is short and nearly constant (Lai et al., 2018; Leija-Martínez et al., 2014; Yoffe et al., 2011).
Our work has several potential extensions.
It is possible that LinearFold can be extended to calculate the partition function and base pair probabilities for natural RNA sequences with well-defined structures, since the classical method for that task, the McCaskill (1990) algorithm, is isomorphic in structure to the cubic-time algorithms that are used as baselines in this article.
This linear-time approach to calculate base pair probabilities should facilitate the linear-time identification of pseudoknots, by either replacing the cubic-time McCaskill algorithm with a linear-time one in those heuristic pseudoknot-prediction programs (Bellaousov and Mathews, 2010; Sato et al., 2011), or linearizing a supercubic-time dynamic program for direct prediction with pseudoknots (Dirks and Pierce, 2003; Reeder and Giegerich, 2004).
We will test the hypothesis that our beams potentially capture cotranscriptional folding with empirical data on cotranscriptional folding (Watters et al., 2016).
Being linear-time, LinearFold also facilitates faster parameter training than the cubic-time CONTRAfold using structured prediction methods (Huang et al., 2012), and we envision a more accurate LinearFold using a model tailored to its own search.
Author contributions
L.H. conceived the idea and directed the project. L.H., D.D. and K.Z. designed algorithms. L.H. and D.D. wrote a Python prototype, and K.Z., D.D. and H.Z. wrote the fast C++ version. D.H.M. and D.H. guided the evaluation that H.Z. and D.D. carried out. L.H., D.D. and H.Z. wrote the manuscript; D.H.M. and D.H. revised it. K.L. made the webserver.
Acknowledgements
We would like to thank the reviewers for suggestions, Rhiju Das for encouragement and early adoption of LinearFold into the EteRNA game, James Cross for help in algorithm design, Juneki Hong and Liang Zhang for proofreading.
Funding
This project was supported in part by National Science Foundation (IIS-1656051 and IIS-1817231 to L.H.), National Institutes of Health (R56 AG053460 and R21 AG052950 to D.H., and R01 GM076485 to D.H.M.).
Conflict of Interest: none declared.
References
Author notes
He Zhang and Dezhong Deng authors wish it to be known that these authors contributed equally.
Present address: Google, Inc, New York, NY 10011, USA.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}