





Abstract
Protein post-translational modifications (PTMs) regulate a wide range of cellular protein functions. Many PTM sites from the same (intra) or different (inter) proteins often cooperate with each other to perform a function, which is defined as PTM cross-talk. PTM cross-talk within proteins attracted great attentions in the past a few years. However, the inter-protein PTM cross-talk is largely under studied due to its large protein pair space and lack of a gold standard dataset, even though the PTM interplay between proteins is a key element in cell signaling and regulatory networks.
In this study, 199 inter-protein PTM cross-talk pairs in 82 pairs of human proteins were collected from literature, which to our knowledge is the first effort in compiling such dataset. By comparing with background PTM pairs from the same protein pairs, we found that inter-protein cross-talk PTM pairs have higher sequence co-evolution at both PTM residue and motif levels. Also, we found that cross-talk PTMs have higher co-modification across multiple species and 88 human tissues or conditions. Furthermore, we showed that these features are predictive for PTM cross-talk between proteins, and applied a random forest model to integrate these features with achieving an area under the receiver operating characteristic curve of 0.81 in 10-fold cross-validation, prevailing over using any single feature alone. Therefore, this method would be a valuable tool to identify inter-protein PTM cross-talk at proteome-wide scale.
A web server for prioritization of both intra- and inter-protein PTM cross-talk candidates is at http://bioinfo.bjmu.edu.cn/ptm-x/. Python code for local computer is also freely available at https://github.com/huangyh09/PTM-X.
Supplementary data are available at Bioinformatics online.
1 Introduction
Post-translational modifications (PTMs) of proteins add another layer to the complexity of the proteome, by reversibly modifying amino acid residues with chemical groups, e.g. phosphate. Recent advances in mass spectrometry have enabled PTMs measurement in a high-throughput manner (Witze et al., 2007), and consequently increased our understanding the biological functions of PTMs (Beltrao et al., 2012; Li et al., 2013) in cellular localization of protein, protein complex formation, etc. In addition, PTMs often interplay with each other to modulate cell signaling and biological processes, known as PTM cross-talk (Beltrao et al., 2013), which has shown many important impacts, including in DNA repair (Ivanov et al., 2007), gene expression regulation (Khidekel and Hsieh-Wilson, 2004) and protein stability (Bengoechea-Alonso and Ericsson, 2009; Esteve et al., 2011; Ruan et al., 2013). One typical example is that more than 10 PTMs on TP53 function cooperatively and precisely in suppressing tumorigenesis (Dai and Gu, 2010). Furthermore, the PTM cross-talk not only occurs within one protein but also between two different proteins, though the latter is normally more difficult to identify due to a larger examination space. As an example of inter-protein PTM cross-talk, the methyltransferase activity of histone-lysine N-methyltransferase EZH2 can be suppressed when Ser-21 of EZH2 is phosphorylated by Akt, which results in a decrease of lys-27 trimethylation on histone H3 (Cha et al., 2005). Also, on tyrosine-protein phosphatase non-receptor type 12, the phosphorylation at Ser-19 changes its substrate interface, which leads to the suppression of activity toward human epidermal growth factor receptor-2 Tyr-1196 site (Li et al., 2018).
Despite the important biological function of PTM cross-talk, its experimental identification is a bottleneck challenge, and has only been applied to small or medium scales, e.g. the global interaction between phosphorylation and ubiquitylation in Saccharomyces cerevisiae (Swaney et al., 2013). On the other hand, computational methods emerge to prioritize PTM cross-talk candidates at whole proteome scale, and much efforts have been made on deciphering the properties of PTM cross-talk from different dimensions, e.g. sequence co-evolution across eukaryotes (Minguez et al., 2012), co-existence of histone modifications in different experiments (Schwammle et al., 2014) and sequence motif pattern for proximate PTMs (Peng et al., 2014). In our previous study (Huang et al., 2015), we found that intra-protein PTM cross-talk can be well predicted by integrating distance, disordered region location and co-evolution information. Very recently, the focus has also been extended to inter-protein level. For example, we found that co-occurrence in multiple tissues or multiple experimental conditions is a good measurement of PTM cross-talk, both within and between proteins (Li et al., 2017). Also, PTMcode v2 extends the PTM cross-talk prediction to inter-protein level by either sequence co-evolution or the physical distance within a complex (Minguez et al., 2015). However, a quality dataset of inter-protein PTM cross-talk is highly demanded to evaluate these predictions, and a set of predictive features remains to be determined, together with development of a powerful classifier to predict PTM cross-talk between proteins.
In this study, we systematically surveyed the published literature to collect experimentally validated inter-protein PTM cross-talk pairs. In total, 199 pairs of PTM sites in 82 pairs of human proteins with experimental support were manually compiled from the published literature. We measured the evolutionary correlations of cross-talk pairs at both sequence and modification levels, including the sequence co-evolution on PTM residues across multiple species and their surround motifs and the PTM co-modification across multiple species and multiple conditions. Except the co-modification across species, the other three features were then integrated into a random forest (RF) classifier to predict PTM cross-talk, achieving an area under the receiver operating characteristic (ROC) curve of 0.81 in 10-fold cross-validation, superior to any single feature alone.
2 Materials and methods
2.1 Cross-talk data collection
We retrieved inter-protein PTM cross-talk pairs from the published literature. We manually reviewed 4067 related articles which were extracted from PubMed on September 21, 2017 with the keywords ‘(residue-specific OR site-specific) AND (cross-talk OR phosphorylation OR acetylation OR methylation OR ubiquitination OR SUMOylation OR O-N-acetylgalactosamine OR O-N-acetylgucosamine)’. In addition, we quarried physically connected PTM pairs between two interactive proteins in PepCyber: P ∼Pep (Gong et al., 2008) (http://www.pepcyber.org/PPEP/), a database of phosphoprotein-binding domains mediated human protein–protein interactions. We only consider these items that are supported in literature. In order to ensure the high quality of the collected PTM cross-talk data, we manually reviewed the publications for each item, and checked their protein sequence in UniProt database (The UniProt Consortium, 2017). During the manually reviewed process in UniProt database, we also found additional PTM cross-talk samples. Finally, 199 inter-protein PTM cross-talk samples across 82 human protein pairs were obtained. The summarized PTM types and their interaction is listed in Table 1, and more details about these 199 samples are given in the Supplementary Table S1, including their positions on the protein sequences, modification types, brief descriptions of the cross-talk mechanism and the data source.
The occurrence of the PTM type combinations in compiled PTM cross-talk pairs
| Number | O-GlcNA | SUMO | Acetyl | Methyl | Phospho |
|---|---|---|---|---|---|
| O-GlcNA | 0 | 0 | 0 | 0 | 4 |
| SUMO | — | 0 | 2 | 0 | 12 |
| Acetyl | — | — | 0 | 2 | 22 |
| Methyl | — | — | — | 2 | 5 |
| Phospho | — | — | — | — | 150 |
| Number | O-GlcNA | SUMO | Acetyl | Methyl | Phospho |
|---|---|---|---|---|---|
| O-GlcNA | 0 | 0 | 0 | 0 | 4 |
| SUMO | — | 0 | 2 | 0 | 12 |
| Acetyl | — | — | 0 | 2 | 22 |
| Methyl | — | — | — | 2 | 5 |
| Phospho | — | — | — | — | 150 |
Note: O-GlcNA: O-GlcNAcylation, SUMO: SUMOylation, Acetyl: Acetylation, Methyl: Methylation, Phospho: Phosphorylation.
The occurrence of the PTM type combinations in compiled PTM cross-talk pairs
| Number | O-GlcNA | SUMO | Acetyl | Methyl | Phospho |
|---|---|---|---|---|---|
| O-GlcNA | 0 | 0 | 0 | 0 | 4 |
| SUMO | — | 0 | 2 | 0 | 12 |
| Acetyl | — | — | 0 | 2 | 22 |
| Methyl | — | — | — | 2 | 5 |
| Phospho | — | — | — | — | 150 |
| Number | O-GlcNA | SUMO | Acetyl | Methyl | Phospho |
|---|---|---|---|---|---|
| O-GlcNA | 0 | 0 | 0 | 0 | 4 |
| SUMO | — | 0 | 2 | 0 | 12 |
| Acetyl | — | — | 0 | 2 | 22 |
| Methyl | — | — | — | 2 | 5 |
| Phospho | — | — | — | — | 150 |
Note: O-GlcNA: O-GlcNAcylation, SUMO: SUMOylation, Acetyl: Acetylation, Methyl: Methylation, Phospho: Phosphorylation.
2.2 Generation of control sets
As a reference, 345 877 human PTM items were downloaded from PhosphoSitePlus® database, version date August 1, 2018 (Hornbeck et al., 2015) (www.phosphosite.org), which not only includes phosphorylation, but also acetylation, methylation, ubiquitination, SUMOylation, O-N-acetylgalactosamine, O-N-acetylglucosamine, etc. As mentioned in this database, these PTMs were manually gathered either from published experiments indexed in PubMed or unpublished data generated at the Cell Signaling Technology (http://www.cellsignal.com). We use the PTMs listed in this database for a candidate space, and for generating an evaluation control set, we only consider those PTM pairs from any of the 82 protein pairs in the cross-talk dataset. We further filtered out those PTM pairs whose both PTM sites are included in the cross-talk set, no matter whether they are recorded as a cross-talk pair or in two separate cross-talk events. This procedure gives us a control set of 13 656 PTM pairs in total. Then, we further filtered out those samples with PTM type combination that is not observed in the cross-talk set. Therefore, we have 11 858 control samples for comparison and prediction analysis in this work. It should be noted that there may be some false negatives in the control set due to the incompletion of experimentally annotated of cross-talk events, however, the false positives in prediction may be reduced thanks to a such control set that is generated from functionally connected protein pairs.
2.3 Sequence co-evolution
In order to study the evolution on protein sequences, multiple sequence alignment (MSA) across about 50 vertebrates species including human were downloaded from the vertebrates non-supervised orthologous groups ‘align’ dataset in the eggNOG database v4.5, date April 11, 2015 (Powell et al., 2014). When multiple paralogs from one species are included in an MSA item, only the one with the shortest editing distance to the human homolog reference is used. For studying a pair of MSAs for two proteins, we only keep the shared species for analysis.
Residue co-evolution
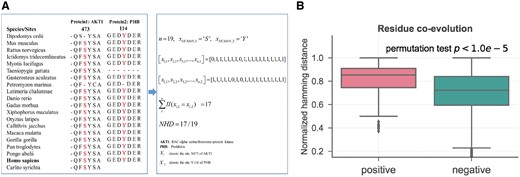
Sequence residue co-evolution analysis of cross-talk PTMs. (A) Demonstration of sequence residue co-evolution with two excerpts of MSA. One excerpt of MSA of protein AKT1 and the other excerpt of MSA of protein PHB across the common 19 species. The two discrete random variables denoting the conservation states of amino acids with 1 for conserved state and 0 otherwise. (B) Comparison of the sequence residue co-evolution scores between the cross-talk set (positive) and control set (negative)
Sequence motif co-evolution
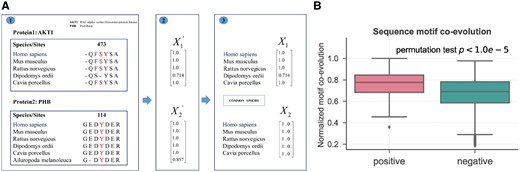
Sequence motif co-evolution analysis of cross-talk PTMs. (A) Demonstration of motif co-evolution with two excerpts of MSA. One excerpt of MSA of protein AKT1 across five species and the other excerpt of MSA of protein PHB across six species. Note that, the two proteins have five common species in the excerpts. (B) Comparison of motif co-evolution scores between cross-talk set (positive) and control set (negative)
2.4 Co-modification across different species and different conditions in human
Co-modification across three species
Co-modification across 88 conditions in human
Besides co-modification across different species, the co-modification across multiple conditions in human is also applied, similar to our previous study (Li et al., 2017). Proteome-wide human phosphorylation dataset measured from 88 different tissues or conditions are used to explore the independence between two phosphorylations. When comparing two PTMs, we can count the number of conditions with both modifications , with a single modification , or , and with no modification . Based on this table of four numbers, we used a Fisher’s exact test to calculate the probability of a null hypothesis that these two modifications happen independently across these 88 conditions. The one-sided P-value with transformation is used as the co-modification score. Therefore, the higher of , the less likely that the two PTMs are independent. Note, we only include those PTMs that have at least one modification state across the 88 conditions for analysis.
2.5 PTM cross-talk prediction and performance evaluation
To integrate different features to predict inter-protein PTM cross-talk, we used the RF model implemented in SciKit-learn (Pedregosa et al., 2011) with the parameter n_estimators of 100 (denote the number of trees in the forest), and the other parameters as default. Since the positive and negative samples are highly imbalanced (199 versus 11 585), we down sampled the negative samples to the size of positive samples and repeated this procedure with the replacement of 100 times. The final prediction result on new samples is the averaged prediction scores from the 100 balanced RF models. We call this ensemble classifier multi-balanced RF (MBRF), and all prediction analyses in this work are based on MBRF. For inter-protein PTM cross-talk, given two PTM sites from different proteins as input, four attributes will be computed: (i) the residue co-evolution, (ii) sequence motif co-evolution, (iii) PTM co-modification across species and (iv) co-modification across different conditions, based on which the probability of cross-talk will be predicted by the MBRF classifier. There are three types of feature combinations for integrative models: (i) both sequence features, (ii) sequence features and modification across conditions and (iii) all four features. It should be noted that the sequence-based features (i.e. residue co-evolution and sequence motif co-evolution) cannot be omitted, otherwise the sample will be skipped for prediction.
We used a 10-fold cross-validation to evaluate the performance of the MBRF prediction models. The cross-talk and control PTM pairs were divided into 10 random equal-sized subsets separately; this split is the same for all feature combinations. Then one subset of the 10 subsets was retained as the test set, and the other nine subsets were used as the training set to build the prediction model. Each of the 10 subsets will be used as the test set once. Note, the down-sample strategy is included in the MBRF rather than the cross-validation split. Due to the small size of the sample set, the random split in the cross-validation may affect the performance evaluation. Therefore, the 10-fold cross-validation process was repeated 100 times by using different 10-fold split, and the prediction results were pooled together to generate an overall ROC curve.
2.6 Permutation test
The standard permutation test is used here to test whether each feature is significantly different from cross-talk set, say A, to control set, say B. As this test does not require any distribution assumption, it is very useful for analyzing some features in this work, particularly the co-modification across species as a categorical feature. Briefly, we calculate the original mean difference d0 between the two sets A and B. Then we pooled A and B together, and randomly re-divided them into two sets with the original sizes as one permutation. Then we can have the mean difference of the permuted two sets for the ith permutation, which is repeated 100 000 times here. Finally, we calculated the two-sided P-value by the proportion of permutations that have .
3 Results
In this study, we manually compiled 199 inter-protein PTM cross-talk pairs from 82 protein pairs across 86 human proteins (see details in Section 2 and Supplementary Table S1). When counting the number of PTM cross-talk events that each protein is involved in (Supplementary Table S2), interestingly we found a few proteins have much more than the majority (median 4 events), especially CDC25C with 26 events, CDK1 with 22 events and AKT1 with 16 events. Also, a few protein pairs have more PTM cross-talk events than others (Supplementary Table S3), e.g. 17 PTM cross-talk events occur between CDC25C and CDK1 as the most. We further present the PTM cross-talk into a protein interaction network (Supplementary Fig. S1), and we surprisingly found that 47 out of the 86 proteins form a sub-graph, suggesting the important roles of PTM cross-talk in cell signaling and regulatory network.
3.1 Sequence co-evolution at residue level and motif level
Sequence co-evolution is widely used to study the functional association between two amino acids, as it presents a conservation interdependence across species in complex ecological networks (de Juan et al., 2013). Here, we explore the sequence co-evolution of inter-protein PTM cross-talk at both a single residue level and a 7-mer motif level.
We first used the NHD to measure how frequent two residues conserve or mutate jointly across around 50 vertebrates. Figure 1A shows an example of AKT1 and prohibitin (PHB) across 20 vertebrates with a cross-talk event between S473 on AKT1 and Y114 on PHB. As described in the Section 2, only the species shared by both proteins are taken into account, thus Carlito syrichta is discarded as it is missing for PHB. For the remaining 19 shared species, 17 species have the same conservation states for both PTM residues (16 co-conserved and 1 co-mutated), which gives a residue co-evolution score of 17/19 for this example. Residue co-evolution scores were further calculated for 168 of the 199 cross-talk pairs, and 8574 of the 11 585 control pairs. The remaining 31 cross-talk and 3011 control pairs do not have this feature because either one of the proteins does not have an MSA or the amino acid of the input PTM does not match the MSA even if one or two position shift is allowed. By comparing the available samples in these two datasets, we found that the cross-talk PTM pairs have a significantly higher residue co-evolution than that of the control PTM pairs (mean: 0.807 versus 0.704, by permutation test, Fig. 1B).
Based on the same MSA data, we extended the sequence co-evolution from the residue level to the sequence motif level. On the same example between protein AKT1 and PHB (Fig. 2A), we first extracted the ±3 amino acids surrounding the PTM sites as a 7-mer motif. For S473 on AKT1, the two residues on −1 and 0 position in Dipodomys ordii was different from their human references, therefore the motif conservation for this species is 5/7 = 0.714. Similarly, we can have the motif conservation scores for all shared species on these two proteins, forming two motif conservation vectors. Then the motif co-evolution score is calculated by taking the dot product between these two motif conservation vectors with normalized to the number of common species. From the same sets of samples as the residue level, i.e. 168 cross-talk pairs and 8574 control pairs, we clearly see that cross-talk PTM pairs also have significantly higher motif co-evolution than that of the control set (mean: 0.754 versus 0.679, by permutation test, Fig. 2B). Together, the two results suggest that sequence co-evolution at both PTM residue level and motif level can be good indicators of PTM cross-talk between proteins.
3.2 Co-modification across different species and different conditions in human
The effectiveness of using protein sequence conservation for analyzing the functional importance of PTMs is possibly because it gives an approximate PTM conservation status across species. Thus, the directly and experimentally verified PTM status across multiple species can be very informative to study the functions of PTM and their interplays (Beltrao et al., 2012; Landry et al., 2009). Indeed, in our previous study (Huang et al., 2015), we have shown that co-conservation of modifications among three species has the potential link to the functional interplay between two PTMs within a protein and can been used to predict intra-protein PTM cross-talk. Here, we apply the co-modification across Homo sapiens, Mus musculus and Rattus norvegicus to measure the modification co-conservation. Same as Huang et al. (2015), the co-modification measures the proportion that the two PTMs conserve simultaneously on the reference residues across the three species. Figure 3A shows example of modification status of two PTM pairs on the proteins AKT1 and PHB in the three species. The cross-talk pair between S473 on AKT1 and Y114 on PHB has co-modification states in human and mouse, giving a co-modification score of 2/3, while the non-cross-talk pair, S475 on AKT1 and S121 on PHB, has co-modification only in human, scoring at 1/3. Even though both PTM pairs have fully co-conserved residues across the three species, the co-modification levels are different, and may imply different functional dependence. Here, for fairness we removed the 13 PTM cross-talk samples whose one or two PTMs are not included in human PTM set in PhosphoSitePlus, and consequently we have 186 cross-talk pairs and 11 585 control pairs for further analysis. By comparing these two sample sets, we found that the score of co-modification across species is significantly higher in cross-talk pairs than that of control pairs (mean: 0.507 versus 0.429, by permutation test, Fig. 3B).
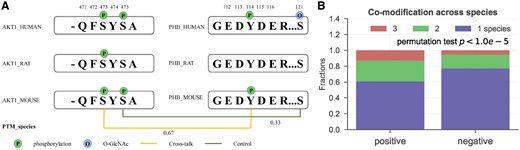
Co-modification across species analysis of cross-talk PTMs. (A) Demonstration of co-modification across species with sequence alignments across human, mouse and rat. (B) Comparison of co-modification across species scores between cross-talk set (positive) and control set (negative)
Besides the evolutionary process, the correlation of modification status across different conditions in one species can also suggest functional associations. In a previous study, we proposed a co-occurrence method to explore functional connections between PTM sites by calculating their tendency to be modified simultaneously across 88 different conditions in human (Li et al., 2017). Here, the same proteome-wide human phosphorylation dataset is used measure the co-modification across conditions for inter-protein PTM pairs (see Section 2 for more details). Figure 4A shows two examples of co-modification across the 88 conditions: a cross-talk sample between Y412 on protein FGR (tyrosine-protein kinase Fgr) and Y281 on SLAF1 (signaling lymphocytic activation molecule), and a control sample between S132 on SHIP2 and Y281 on SLAF1. Their phosphorylation status (red: on, blue: off) across 88 conditions are shown in the heatmap, where we can calculate the co-modification scores, i.e. −log10(p) in Fisher exact test, for these two examples and have 12.549 for cross-talk sample and 0.397 for control sample. As this feature is only available for phosphorylation-phosphorylation pairs, we only have co-modification scores for 87 of 199 cross-talk and 3040 of 11 585 control PTM pairs. Still, we see that the cross-talk pairs show a clearly higher co-modification across multiple conditions than that of the control pairs (mean: 2.111 versus 1.044, by permutation test, Fig. 4B), indicating that the cross-talk PTM pairs have much higher chance to reject the independence null hypothesis than the random PTM pairs. Together, the above two analyses reveal that co-modification across different species and different conditions can be predictive features for identifying inter-protein cross-talk pairs.
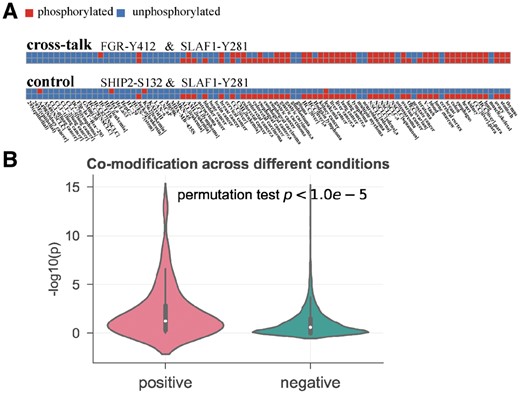
Co-modification across different conditions analysis of cross-talk PTMs. (A) Demonstration of co-modification across 88 conditions for two PTM pairs (all phosphorylations; cross-talk: Y412 on FGR and Y281 on SLAF1; control: S132 on SHIP2 and Y281 on SLAF1, achieved the score of 12.549 and 0.017, respectively). The specific information of 88 conditions is listed in Supplementary Table S2. (B) Comparison of co-modification across different conditions scores between cross-talk set (positive) and control set (negative)
3.3 Integrative prediction of PTM cross-talk between proteins
As demonstrated above, the inter-protein PTM cross-talk pairs display evolutionary correlations at both sequence level and modification level. Therefore, we ask if these four properties can be used to predict PTM cross-talk between proteins. First, we tested the discrimination power of each of the four features by 10-fold cross-validations. The area under the curve (AUC) values in Figure 5A show that the sequence co-evolution on the PTM residue is the most discriminative feature (AUC = 0.785), and it also has a relatively low no-call rate, namely only 31 out of 199 cross-talk and 3011 out of 11 585 control pairs do not have the residue co-evolution measures. Following features are sequence motif co-evolution (168 cross-talk samples, AUC = 0.685) and co-modification across conditions (87 cross-talk samples, AUC = 0.654). By contrast, the performance of co-modification across species was relatively poor (186 cross-talk samples, AUC = 0.558), partly due to the incompleteness of PTM data in mouse and rat. Then, we further ask if the integration of these four features can improve the prediction comparing to using a single feature alone. For fairness, we only used the 76 cross-talk samples and 2593 control samples that have all these four features to compare single-feature models and integrative model. Unsurprisingly, the performance with each single feature alone slightly decreases on this smaller dataset comparing to use all available samples before (see single feature in Fig. 5A and B). However, the integration of three predictive features, i.e. sequence co-evolution and co-modification across conditions, has the best performance and increases the AUC to 0.814 from 0.756 by a single feature alone (i.e. residue co-evolution). Due to the limited prediction power of co-modification across species, this feature fails to improve the performance in the integrative model by adding it. Therefore, we omit this feature in the integrative model.
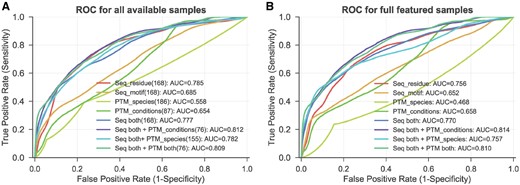
Evaluating the performance of predicting PTM cross-talk using different features combinations; 10-fold cross-validation with repeating 100 times are pooled together to generate an overall ROC curve. (A) Evaluation is performed on all available samples for each feature (combination); the size of cross-talk samples are presented in the brackets. (B) Evaluation is performed on 76 cross-talk and 2593 control fully featured samples. Abbreviations: sequence residue co-evolution (Seq_residue), sequence motif co-evolution (Seq_motif), co-modification across species (PTM_species), co-modification across different conditions (PTM_conditions), both sequence co-evolution (Seq both), both co-modification (PTM both)
Though the co-modification across conditions contributes a lot to the integrative model, a large number of samples do not have this attribute. Therefore, we also recommend the usage of only both sequence co-evolution features for most PTM pair candidates. Also, the sequence feature combination gives more than double cross-talk sample size comparing to that with co-modification across conditions (168 versus 76). Additionally, Figure 5B suggests that in this small sample set, the integration of both residue and motif co-evolution gives better performance than either of them alone, though this improvement is marginal, and need to be examined more extensively.
3.4 Influence of PTM type bias on prediction performance
Among the 199 inter-protein PTM cross-talk pairs, 150 pairs are cross-talk events between two phosphorylation sites (Table 1). In other words, the compiled cross-talk set is bias toward the phosphorylation-phosphorylation PTM types. It is not clear if the prediction model can be used for PTM types that are not included or underrepresented in the training set. To test the influence of PTM types, we trained MBRF models with only phosphorylation-phosphorylation cross-talk pairs (150 cross-talk set pairs and 7312 control pairs), and tested the prediction performance on the rest PTM types (49 cross-talk pairs and 4273 control pairs). Figure 6 shows that phosphorylation–phosphorylation dataset is predictive for other PTM types (AUC = 0.777), even though only two sequence co-evolution features are available. With a threshold of 0.65, the false positive rate can be as low as 9.7% and the true positive rate is 38.5%. This prediction is equivalent as an independent test, evidencing the power of our method in predicting inter-protein PTM cross-talk and its robustness to PTM type bias.
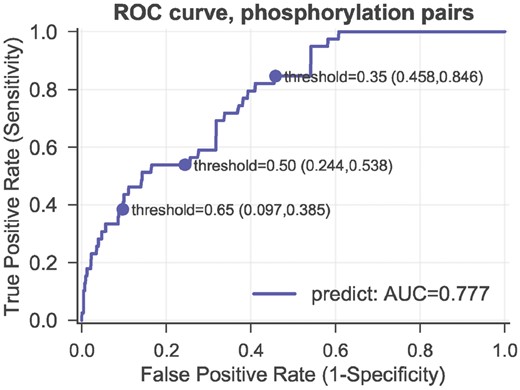
Evaluating the robustness of the prediction model using biased training sets (phosphorylation– phosphorylation dataset). The ROC curves of the MBRF classifier using phosphorylation– phosphorylation dataset as training set and the rest as testing set. The false positive rate and true positive rate are presented in the brackets following the corresponding threshold 0.35, 0.5 and 0.65
3.5 PTM-X online server
Combining our previous intra-protein prediction method, we provide a web server named PTM-X for the prediction of intra- and inter-protein PTM cross-talk (http://bioinfo.bjmu.edu.cn/ptm-x/). The MBRF prediction model in the web site was trained with all human cross-talk and control pairs, for two types of feature combinations: (i) residue and motif sequence co-evolution and (ii) the addition of co-modification across conditions. Users can input candidate PTM pairs by specifying the protein UniProt accession number and the PTM positions on protein sequences. Then PTM-X server will give a final prediction result for each PTM pair by using the same feature combinations, by displaying on the web with a download link to a text file (see example in Supplementary Fig. S3). The input PTM pairs can be taken as potential cross-talk pairs if their prediction scores are higher than a given threshold. Generally, a strict threshold gives lower false positive rate but higher false negatives, while a more lenient threshold can be used to obtain more sensitive predictions. We provide an interface to facilitate this procedure, if users click on the prediction score on the web page, the ROC curve from the 10-fold cross-validation will appear and display the related false positive and true positive rate with the prediction score as a selected threshold (Supplementary Fig. S3).
4 Discussion
In this study, we extended our previous work in intra-protein PTM cross-talk prediction (Huang et al., 2015) to inter-protein level. However, the methods for PTM cross-talk prediction cannot be directly migrated from intra-protein level to inter-protein level. The most discriminative feature for PTM cross-talk within protein is the proximate location on both primary sequences and tertiary structures, which is unavailable for PTM pairs from two proteins. Physical distance within a protein complex can be an alternative measurement for inter-protein PTM pairs (Minguez et al., 2015), while this information is only available for a very small subset of protein pairs and will result in a large number of missing values when used for prediction (all missing for cross-talk and 99.7% missing for control).
On the other hand, the protein sequences are much easier to access, therefore, the sequence co-evolution was first extended to inter-protein level for functional association between PTM sites (Minguez et al., 2012). Initially, mutual information (MI) and its normalized value (nMI) were successfully applied to measure this feature at the intra-protein level (Huang et al., 2015; Minguez et al., 2012). However, we noticed that nMI is not suitable when extending from intra-protein to inter-protein level (Supplementary Fig. S4). One possible reason is that inter-protein PTM pairs have much higher sequence divergence than intra-protein counterpart, but nMI is very sensitive to this divergence and exponentially decreases to zero (Supplementary Fig. S4C–F). Therefore, in this work we applied the NHD to measure sequence co-evolution, which shows clearly better prediction performances than nMI (Fig. 5A and Supplementary Fig. S4B).
Though the sequence co-evolution across vertebrates has been shown a very predictive feature for PTM cross-talk, it is less certain whether there is an optimal range of species that sequence co-evolution predicts PTM cross-talk best. When stretching from vertebrates to animals, the sequence co-evolution decreases for both positive and negative samples, which has a strong correlation with the number of species or the maximum evolutionary distance in the animal set (Supplementary Fig. S5C–F). Surprisingly, cross-talk PTMs decrease more severely than non-cross-talk PTM pairs (Supplementary Fig. S5C–F). After further extending to eukaryotes and all organisms in eggNOG, cross-talk PTMs even nearly lose its advantage on this dimension and show no clear difference to the background PTM pairs (Supplementary Fig. S6A for NHD and Supplementary Fig. S7A for nMI). This reduction of the prediction power remains even if we down sampled the negatives to achieve the same distribution of species numbers between positive and negative sets (Supplementary Figs S6B and S7B) or when we removed protein pairs with too high or too low number of species (Supplementary Figs S6C and S7C). Together, these observations imply that an optimal evolutionary range may exist for cross-talk PTMs to achieve highest gain in sequence co-evolution comparing to background PTM pairs, but admittedly a larger set of PTM cross-talk samples are needed to enhance this hypothesis.
Furthermore, the most precious though still limited data is the direct measurement of PTMs in different species and different conditions, both of which have been shown good potential in predicting PTM cross-talk within protein (Huang et al., 2015; Li et al., 2017). Here, again we evidenced that cross-talk PTM pairs between proteins also have higher co-modification across three species and multiple conditions. We expect that the co-modification features have a great potential in predicting PTM cross-talk, however due to incompleteness, only three species, human, mouse and rat, are used in this work (see Supplementary Table S5). Other species, including Saccharomyces, Arabidopsis, Caenorhabditis and Drosophila, have 2000–5000 verified PTMs, but they are relatively far from human and hence have much fewer orthologs comparing to mouse or rat (Supplementary Table S6). Alternatively, using predicted PTM data, e.g. dbPTM (Huang et al., 2016), which is better covered, may relieve this issue, though the ideal way is to coherently model the PTM status and their cross-talk.
We further applied a RF classifier to predict PTM cross-talk between proteins by integrating three predictive features: residue and motif co-evolution, co-modification across different conditions. The co-modification across species is not included due to its limited prediction power, though it shows a good potential but probably requires more completed PTM sets. Still, the integrative model achieves good prediction and outperforms any single feature alone. In order to reduce the no-call rate in the integrative model, it is also sensible to omit the co-modification across conditions and use the two sequence co-evolution features only, which balance the prediction performance and the usage of the samples. Furthermore, we saw that the model trained by phosphorylation-phosphorylation cross-talk subset can predict well on other PTM types, indicating that the prediction model can be used to unseen or less represented PTM types.
Though we believe PTM-X offers a valuable new tool to prioritize a large number of inter-protein PTM cross-talk candidates, there are also many other directions for investigation. First, a bigger dataset of validated inter-protein PTM cross-talk events is still highly demanded, through which more sophisticated model can be applied to achieve better prediction performances, and distinct properties of cross-talk events may be revealed between different PTM types. Second, more information can be integrated into the prediction model, e.g. the protein level interaction may increase the baseline in prediction of the interaction between PTMs, by filtering PTMs from less interactive protein pairs (Minguez et al., 2015). Third, the predicted PTM cross-talk between proteins may in return improve our understanding of protein or gene regulatory network; the big sub-graph among the 199 cross-talk samples is a very interesting example (Supplementary Fig. S1).
Funding
This work was supported by the National Key Research and Development Program of China [2018YFA0507504]; the National Natural Science Foundation of China [61773025]; and the Interdisciplinary Medicine Seed Fund of Peking University [BMU2017MB001].
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Rongting Huang, Yuanhua Huang and Yubin Guo authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}