





Abstract
Deep mutational scanning experiments have enabled the measurement of the sequence-function relationship for thousands of mutations in a single experiment. The Protein Analysis and Classifier Toolkit (PACT) is a Python software package that marries the fitness metric of a given mutation within these experiments to sequence and structural features enabling downstream analyses. PACT enables the easy development of user sharable protocols for custom deep mutational scanning experiments as all code is modular and reusable between protocols. Protocols for mutational libraries with single or multiple mutations are included. To exemplify its utility, PACT assessed two deep mutational scanning datasets that measured the tradeoff of enzyme activity and enzyme stability.
PACT efficiently evaluated classifiers that predict protein mutant function tested on deep mutational scanning screens. We found that the classifiers with the lowest false positive and highest true positive rate assesses sequence homology, contact number and if mutation involves proline.
PACT and the processed datasets are distributed freely under the terms of the GPL-3 license. The source code is available at GitHub (https://github.com/JKlesmith/PACT).
Supplementary data are available at Bioinformatics online.
1 Introduction
Deep mutational scanning (DMS) is a powerful method to assess the function of thousands of protein genotypes in a massively parallel fashion (Fowler et al., 2010). These experiments are set up such that naïve unselected population sequences are compared to a population with a particular function after a screen or selection. The per-mutant change of frequency upon selection is computed, which is a quantitative readout for the selected function. DMS has been applied to many diverse protein systems involved with binding or epitope mapping (Wang et al., 2017; Whitehead et al., 2012), enzyme catalysis or metabolic pathway flux (Kotler et al., 2018; Romero et al., 2015; Stiffler et al., 2015), viral inhibition (Ashenberg et al., 2017), protease susceptibility (Rocklin et al., 2017) and chaperone engineering (Medina‐Cucurella Angélica et al., 2018). These experiments typically involve comprehensive site saturation libraries, which diversify each residue to all 20 amino acids and nonsense codons. Recent methods have enabled the creation of these genetic libraries in rapid and high-throughput manner (Firnberg and Ostermeier, 2012; Wrenbeck et al., 2016). Future experiments will start to utilize deeper multi-mutant libraries given the development of mass-produced low cost oligo pools (Klein et al., 2016). Furthermore, data repositories like ProtaBank (Wang et al., 2018) are making DMS data readily available for analysis.
In this work, we describe a new software package, Protein Analysis and Classifier Toolkit (PACT), which provides workflows to apply sequence and structural analyses to deep sequencing datasets from DMS experiments (Fig. 1). All analyses are performed by user-shareable protocols that can be customized for any experimental workflow. Furthermore, it is possible to use some of the included analyses on non-DMS datasets. PACT includes a protocol to process deep sequencing reads to per-mutation fitness metric values or import existing mutational datasets calculated from external packages like Enrich (Fowler et al., 2011; Rubin et al., 2017) or dms_tools (Bloom, 2015). The PACT fitness protocol replicates the core functionality of these existing packages with some improvements and differences; however, PACT goes beyond their scope (Fig. 1). In short, existing software packages process deep sequencing reads then calculate a metric of per-variant function. For example, Enrich2 provides an implementation of a random-effects model that is geared for calculating variant enrichment over multiple time points while combining multiple replicates. Experiments targeted by the PACT fitness protocol are end-point with a reference library and a selected library where replicates are processed separately and compared via correlation statistics. More importantly, other protocols within the PACT platform are not included within existing packages. PACT protocols include routines to (i) create, import and combine fitness metric datasets, (ii) compare datasets against each other and against sequence or structural measurements and (iii) perform statistical analyses. PACT protocols (Fig. 1) include:
![Relationship of PACT protocols to features, and comparison of scope of existing packages. (A) PACT offers a protocol (fitness) to process DMS deep sequencing data and calculate the per-mutation fitness metrics for comprehensive single or multiple point libraries. This functionality is replicated in various formats by Enrich, dms_tools and other existing packages. The per-mutation fitness metric data from the fitness protocol or outside packages is imported and made available for the analysis protocols. Analysis protocols serve three functions: (i) assess the DMS datasets against other DMS datasets or versus sequence or structural features, (ii) provide a direct method to calculate and output feature measurements and (iii) combine DMS datasets with features to train classifiers or assess datasets against existing classifiers. Analysis protocols leverage the variance of synonymous codon fitness metrics (Classification) to classify the function of non-synonymous mutations relative to wild-type. This data is combined with features to provide insight into the effect of mutations on function, and enable statistical classifier training [inset: the structure of levoglucosan kinase (PDB: 4ZLU) with residue G359 highlighted and the probability of mutational class versus distance to active site]. (B) Scope of existing DMS tools versus PACT](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bioinformatics/35/16/10.1093_bioinformatics_bty1042/4/m_bty1042f1.jpeg?Expires=1716404802&Signature=2WiDvtEyFdnyoIxwGCqgixsj0wP2Hl6MYnM9Tx0TS89dQAjvPyv9XO5cQ6YqqQrNZwapAco5gO2LwgOqiqpkcEjP2nw1vO8aZHILXBZralFHaIQ0vTcs6MJw-Ysk1JcmqTFvTDSXTERtRuSiXX5t4kbZy9stkV-W4DDGsRy6LaWkFPW9n5HyJ5aMWQstZtDx-sSixLyBNJ7k8rmAHdbJbCXCsgb~mPkEIekKoixGxztALZPW6PUcbOIqiqkmJlhTMqQFfUwelIUNtkEYyTQ-59ZSsflNxGVKITpCQVCqUX1mgM5dl87lVh2ynadvhSGXppBQNHFmdBaOaez3AWO8rg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Relationship of PACT protocols to features, and comparison of scope of existing packages. (A) PACT offers a protocol (fitness) to process DMS deep sequencing data and calculate the per-mutation fitness metrics for comprehensive single or multiple point libraries. This functionality is replicated in various formats by Enrich, dms_tools and other existing packages. The per-mutation fitness metric data from the fitness protocol or outside packages is imported and made available for the analysis protocols. Analysis protocols serve three functions: (i) assess the DMS datasets against other DMS datasets or versus sequence or structural features, (ii) provide a direct method to calculate and output feature measurements and (iii) combine DMS datasets with features to train classifiers or assess datasets against existing classifiers. Analysis protocols leverage the variance of synonymous codon fitness metrics (Classification) to classify the function of non-synonymous mutations relative to wild-type. This data is combined with features to provide insight into the effect of mutations on function, and enable statistical classifier training [inset: the structure of levoglucosan kinase (PDB: 4ZLU) with residue G359 highlighted and the probability of mutational class versus distance to active site]. (B) Scope of existing DMS tools versus PACT
fitness: the per-variant fitness metric values are calculated from deep sequencing. Mutation fitness metric data are saved as a heatmap, column dataset and as a binary encoded Python dictionary file of the entire dataset. Mutation combinations that are mutually beneficial in multi-site libraries are calculated (Dunn et al., 2008). A brief comparison to existing software packages is also included (Note S1).
classification_features: mutations from PACT fitness datasets are classified based on z-score or fitness metric values then all sequence and structural features are calculated and combined as a training dataset for used within other protocols (Note S2).
function_filter: fitness metrics, sequence and structural features are binned and counted. These same mutations can be scored against a naïve Bayes classifier trained on the enzyme levoglucosan kinase (LGK)-WT/LGK.1 and binary filtering (Note S3).
sequence_homology: mutations are compared to homologous sequence site-wise frequency or PSIBlast Position-Specific Scoring Matrix (PSSM) data (Goldenzweig et al., 2016) (Note S4).
structure_analysis: the distance to active site, contact number, distance to surface, distance to interface, (relative) accessible surface area and fraction burial per residue are calculated for a PDB input (Note S5).
Shannon_entropy: site-wise Shannon entropy (Kowalsky et al., 2015) is calculated from enrichment values (Note S6).
back_to_consensus: the probability of mutation type at consensus or non-consensus residues is calculated using site-wise homologous sequence frequency or PSIBlast PSSM data (Note S7).
pact_vs_pact: mutations from fitness datasets are compared against each other or against available features (Note S8).
pact_vs_feature: mutations from a fitness dataset are compared against various features (Note S9).
tools: additional tools for library creation and FASTQ processing are accessible via the protocol and the command line (Note S10). Examples are routines to import csv files of mutational fitness metric data for analysis protocols, split FASTQ files based on shared start sequence and creation of single-site saturation primers.
We describe how PACT processes deep sequencing reads into per-mutation fitness metrics for single-site and multi-site amino acid libraries. We then detail protocols that apply sequence and structural analyses to processed deep sequencing datasets. Finally, to show the utility of PACT we examined published comprehensive datasets that selected for mutations that enhance stability or activity of the enzyme LGK to train a naïve Bayes classifier and binary filters to identify mutations that enhance stability and retain activity via numerous sequence and structural features. We then test this classifier and filter against a separate enzyme, amidase AmiE. We found that the filter with the lowest false positive and highest true positive rate assesses sequence homology, contact number and if the mutation involves proline.
2 Implementation
The minimum runtime requirement is 64-bit Python version 3.4. PACT is operating system independent and has been tested on Windows, OSX, Linux and on a distributed Linux cluster. Recommended hardware is a multi-core processor with 8 GB of RAM. PACT primarily uses the standard Python library to allow greater system compatibility; however, some external Python packages and external programs are required for various protocols. Required packages are NumPy and SciPy for mathematical operations and matplotlib for figure generation. External software utilized by individual features and protocols and instructions to link to these programs is in Note S11. Individual protocols are Python scripts that constitute analysis workflows. The main script pact.py loads a user-defined config text file. This file selects the specific protocol and provides user customizable options for the protocol. Protocol workflow descriptions, config file options, inputs and outputs are explained in Notes S1–S10.
The calculated metric of mutational function in many DMS experiments is the log2 enrichment of the frequency change of a variant in the selected (or final) population relative to the reference (or initial) population (Note S12). Available fitness metrics offered by the fitness protocol are in Supplementary Table S1 with extended derivation in Note S12. Also, in experiments where gene length exceeds sequencing read length, gene sections (called ‘tiles’) are independently mutated, screened and sequenced. This approach necessitates the use of fitness metrics that normalize the variant enrichment using internal and experimental measurements to enable cross-tile comparisons, which PACT efficiently accomplishes.
One source of error in DMS experiments is in the frequency calculation of each library member. This error can be modeled as a Poisson process as library members with low counts from depletion or low abundance have larger errors versus library members with a larger representation (Supplementary Fig. S1). A minimum read count threshold in either the reference or selected population is enforced. For variants with no counts in a population and significant counts in the other, a conservative count of 1 is added for that variant to enable calculation of the log2 enrichment. Because the minimum error with counting approximates Poisson noise, PACT calculates the fitness metric variance through propagation of errors (Klesmith et al., 2015) (Note S12).
To quantify the strength of depletion or enrichment, mutations are compared to neutral variance, which is approximated from a Gaussian distribution (Hietpas et al., 2011; Klesmith et al., 2017) of synonymous wild-type genes (i.e. silent genetic mutants). Per-variant depletions or enrichments are reported as the number of synonymous standard deviations from wild-type (z-score), and evaluated for statistical significance using Welch’s t-test. Alternately, analysis protocols will accept user-defined fitness metrics instead of z-score to classify mutations. The sample size for any given non-synonymous variant is approximated by a calculated expectation value for the number of experiments performed on the variant within the screen for FACS (Note S13) or for growth (Note S14).
3 Identification of classifiers that predict the function of mutations
To demonstrate the utility of the PACT platform, we examined published comprehensive DMS datasets from the enzymes LGK (Klesmith et al., 2015), and the amidase AmiE (Wrenbeck et al., 2017) to discover classifiers using combinations of sequence and structural features that identify mutations beneficial for activity and stability with a low false positive rate of deleterious mutations. We focused on the enzyme LGK as it has two comprehensive datasets, one starting from the wild-type sequence (LGK-WT) and a second starting with a more stable yet similarly active triple mutant (LGK.1). These DMS datasets were complemented with experimental enzymatic activity and stability measurements for numerous purified isogenic single-point variants. As the experimental selection temperature was above the Tm of LGK-WT, enzyme activity assays of purified isogenic single-point mutants indicated that the majority of mutations enriched within that selection had increased thermal stability with a concurrent decrease of enzymatic activity. This result was opposite of LGK.1 as the starting enzyme Tm was above the selection temperature; therefore mutations enriched in this selection primarily enhanced enzyme activity. The combined analysis of these two datasets provides a view into the role of mutations that trade-off enzyme stability and enzyme activity and form our training set to predict function.
Our rationale for the current work originates from a recent publication that leveraged the LGK.1 enzyme activity dataset in combination with a yeast surface display solubility screen to identify solubility-enhancing mutations that do not hinder activity (Klesmith et al., 2017). The term solubility referred to the probability that a protein is properly folded upon translation, which is a function of protein aggregation propensity, thermodynamic stability and folding rate, among other factors. This experimental screen combined with computational sequence and structural features trained on the LGK.1 dataset led to the creation of a binary mutation filter that is able to identify and remove deleterious mutations with a 3% false positive rate. Herein, we assessed if it would be possible to predict the function of mutations via purely computational methods without using data informed from an experimental solubility screen while retaining a similar false positive rate.
We processed the LGK-WT and LGK.1 deep sequencing stability/activity datasets using the fitness protocol with the growth fitness metric. Library coverage and wild-type synonymous statistics are in Supplementary Table S2. We approximated neutral mutations as those within 1.5 SD of wild-type synonymous mutations. The average library fitness metric for both selections is ∼0.16 at 1.5 SD, which correlates to a 12% increase (+1.5 SD) or 10% decrease (−1.5 SD) in the growth rate of a library variant relative to wild-type (Supplementary Fig. S2). Within this range, mutations are considered neutral, while growth above 12% is beneficial and below −10% is deleterious. Comparing the LGK-WT and LGK.1 DMS datasets allows mutations to be classified into nine different stability and activity categories based on individual variant z-scores (Supplementary Fig. S3). To validate the classification prediction categories, we assessed if enzymatic activity and stability measurements from the 15 published isogenic LGK single-point mutation variants (Klesmith et al., 2015) matched the functional prediction of the category that they were classified in. Ten out of 15 (67%) matched our expectations where the measured change in stability or activity matched the cross-comparison categories (Supplementary Table S3). Of the five that did not match our expectations, three had higher activity and one had higher stability than predicted.
Previous work has shown that mutations to any residue seen in protein homologs reduced the rate of a deleterious mutation on function as compared to the basal selection rate (Cochran et al., 2006). We used the consensus protocol to assess mutations to the consensus residue or any residue observed in sequence homologs at sites where the wild-type was not the consensus (Supplementary Fig. S4). The basal rate for deleterious mutation in LGK.1 is 78% whereas any mutation observed in sequence homologs dropped the rate to 67%. If we limit mutations to sites for which wild-type was not consensus, mutating to any observed homolog mutation or the consensus dropped the deleterious rates to 55 and 39%, respectively. Furthermore, if we constrain this analysis to surface residues (Supplementary Fig. S5), the rate of deleterious mutations drops to 23%.
We calculated naïve Bayesian classification probabilities from the LGK-WT and LGK.1 datasets for the newly developed consensus features (Supplementary Figs S4 and S5) with other sequence and structural features (distance to active site, contact number, and PSSM, fraction burial of a residue, and the size and chemical change of mutation based on distance to active site, contact number and fraction burial). Previous work indicated that residue size and chemical change did not show power in classification (except mutations to or from proline) (Klesmith et al., 2017). However, we hypothesize that if we bin the mutation types based on burial or location we could potentially resolve mutation classifications. We reduced the nine LGK-WT/LGK.1 mutation categories into just three: (i) beneficial z-score >1.5 on the LGK.1 selection or >1.5 on the LGK-WT and within ±1.5 on LGK.1 (as these should be desirable active stabilizing mutations); (ii) neutral on LGK.1 and non-beneficial on LGK-WT and (iii) deleterious on LGK.1. Feature counts used for probability generation are in Supplementary Table S4.
To assess generalizability of the naïve Bayes classifier we assessed all combinations of LGK feature Bayesian probabilities against published comprehensive fitness metric data of the amidase enzyme AmiE (Wrenbeck et al., 2017) selected on the two substrates acetamide (Fig. 2) and propionamide (Supplementary Fig. S6). We approximated AmiE neutral mutations if they had a fitness metric that was within ±0.15 or ∼ ±10% change in growth rate which is similar to the LGK z-score classification threshold. Single selection basal rates are listed in Supplementary Table S5. We based our comparisons on the predicted combination of beneficial and neutral mutations as there are significantly more truly beneficial mutations predicted as neutral than beneficial (Supplementary Fig. S7). The frequency of identifying a beneficial mutation versus a deleterious mutations indicated a group of classifier combinations that were putatively higher performing (Fig. 2A and B, Supplementary Fig. S6 and Tables S6 and S7). This group was also evident if the fraction of deleterious mutations and fraction of beneficial mutations were compared between the acetamide and propionamide datasets (Fig. 2B, Supplementary Fig. S6B).
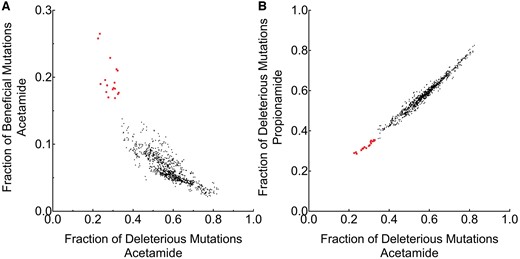
Bayesian classifier optimization indicates a group of feature combinations with increased predictive power. The predictive performance of all 210 combinations of feature Bayesian probabilities trained on LGK datasets (Supplementary Table S4) was evaluated on fitness data from the AmiE amidase datasets. Each dot is a combination of feature Bayesian probabilities for mutations predicted to be beneficial or neutral in activity. A subset of feature Bayesian probabilities (red color, squares; Supplementary Tables S6–S8 and Fig. S6) exhibited a high rate of finding a truly beneficial mutation for AmiE activity on acetamide (A), and a low rate of finding a truly deleterious mutation between acetamide and propionamide (B) if the mutation was predicted to be beneficial or neutral. This subset shows similar performance characteristics between the acetamide and propionamide selections. The best Bayesian classifier assesses if the wild-type residue is consensus, if the mutation is consensus, and the natural wild-type homolog frequency
The best Bayesian classifier—with the lowest deleterious false positive rate and the maximum beneficial mutations per deleterious mutation found—assesses if the wild-type residue is consensus, if the mutation is consensus, and the natural wild-type homolog frequency. About 19% of predicted beneficial mutations are actually deleterious (Supplementary Table S7). Other top features in this group included the natural homolog frequency of the mutation, and contact number binned on proline mutations (Supplementary Table S6).
We then wanted to compare this classifier to (i) the computational portion of the previously published (Klesmith et al., 2017) binary filter: PSSM ≥0, distance to active site ≥15 Å, contact number ≤16 and excluding any proline-involved mutations (‘old filter’); and (ii) this filter plus features based on the herein research: fraction burial <0.4 and at sites where wild-type is not consensus (‘new filter’). For the AmiE acetamide dataset the false positive rate of the old and new filters were 58 and 31%, respectively. Similarly, for LGK the false positive rate for the old and new filters is 35 and 9.9%, respectively (Supplementary Table S8).
An alternate way to assess these filtering approaches is to judge them upon their expected functional change in growth rate. The previous work classified mutations based on the expected phenotype by binning mutations as neutral if their fitness metric values were expected to be ≥80% of the growth rate of wild-type, slightly deleterious if they were <80 and ≥50% of the wild-type growth rate and deleterious if they were <50% of the wild-type growth rate (Klesmith et al., 2017). Doing so significantly lowers the basal deleterious rate of each selection (48 versus 88% for AmiE acetamide, and 34 versus 78% for LGK.1), however, the corresponding false positive rate also decreased and is similar to or better than the reported value from the combined yeast surface display and computational filter screen of 3% for LGK (Supplementary Tables S5 and S8). The deleterious false positive rate for the AmiE acetamide dataset was 0.0% for the Bayesian classifier while the old and new binary filters were 9.1 and 1.8%, respectively. The resulting false positive rate was 3.6 and 1.4% for the old and new filter, respectively, for LGK (Supplementary Table S7).
4 Discussion
We built PACT as a software package to aid in the processing and analysis of DMS experimental datasets. We demonstrated the utility of processing comprehensive DMS experiments and analysis of fitness metric information to train and build mutational filters that identify mutations deleterious for enzymatic function. We trained a naïve Bayesian classifier on published datasets from the enzyme LGK and assessed generalizability on datasets from the enzyme AmiE. We identified that classifiers selective for excluding deleterious mutations utilize features based on sequence homology and contact number binned on proline mutations.
In the most stringent case for the acetamide substrate, the rate of finding a deleterious mutation via the naïve Bayesian classifier outperformed the updated binary filter (19 versus 31%) in our test dataset. While 19% is not perfect, it is an improvement over the basal rate of 88%. In practical application, the 19% deleterious rate over the 88% basal rate would lead to 66 less deleterious variants per 96 tested. In addition, our solely computational filters are comparable or better than the previously published combination experimental yeast surface display/computational filter indicating that a separate experimental screen is not needed if there is sufficient sequence and structural homology data available. However, a separate experimental screen may still be needed depending on the context of where the protein is being tested. This may be the case where certain residues are highly optimized for a given organism, but are sub-optimal in the context of other organisms. Sequence homology would then not be the best feature.
Acknowledgements
We thank M. Faber, A. Golinski and E. Wrenbeck for testing and T. Whitehead for comments on a draft of this manuscript.
Funding
This work was supported by the National Institutes of Health [GM121777 to B.J.H.].
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}