





Abstract
Experimental structure probing data has been shown to improve thermodynamics-based RNA secondary structure prediction. To this end, chemical reactivity information (as provided e.g. by SHAPE) is incorporated, which encodes whether or not individual nucleotides are involved in intra-molecular structure. Since inter-molecular RNA–RNA interactions are often confined to unpaired RNA regions, SHAPE data is even more promising to improve interaction prediction. Here, we show how such experimental data can be incorporated seamlessly into accessibility-based RNA–RNA interaction prediction approaches, as implemented in IntaRNA. This is possible via the computation and use of unpaired probabilities that incorporate the structure probing information. We show that experimental SHAPE data can significantly improve RNA–RNA interaction prediction. We evaluate our approach by investigating interactions of a spliceosomal U1 snRNA transcript with its target splice sites. When SHAPE data is incorporated, known target sites are predicted with increased precision and specificity.
Supplementary data are available at Bioinformatics online.
The function of many if not most non-coding (nc)RNA molecules is to act as platforms for inter-molecular interaction, which depends on their structure and sequence. A large number of ncRNAs regulate their target RNA molecules via base-pairing. For instance, small (s)RNAs regulate the translation of their target genes by direct RNA–RNA interactions with the respective messenger (m)RNAs (Backofen and Hess, 2010). To predict such interactions, regions not involved in intra-molecular base pairing have to be identified. This ‘free-to-interact’ potential of a region, i.e. its unpaired probability, is computed by assessing the fraction of structures where the region is free (unpaired) within the overall structure ensemble of an RNA [(Raden et al., 2018) for a detailed introduction]. The probability are used by state-of-the-art prediction tools like IntaRNA (Mann et al., 2017) to account for the regions’ accessibility. While correct within their thermodynamic models, such probabilities do not incorporate all cellular constraints and dynamics that define accessible regions and thus the likelihood for interaction.
The accuracy of RNA structure prediction improves when experimental structure probing data, such as SHAPE (Wilkinson et al., 2006), is incorporated (Hajdin et al., 2013). This is done by converting the chemically sensed reactivity values to pseudo-energy terms. Pseudo-energies are combined with structure formation energies from thermodynamic models, that are used for RNA structure prediction (Lorenz et al., 2016; Montaseri et al., 2017; Spasic et al., 2018). As SHAPE [For simplicity, SHAPE refers to any structure probing experiment (e.g. SHAPE, DMS)] reactivity is associated with the accessibility of nucleotides, the use of such experimental data is even more promising to improve the accuracy of RNA–RNA interaction prediction methods. For that reason, we introduce a seamless incorporation of SHAPE data into accessibility-based prediction approaches such as IntaRNA.
Recently, structure probing has been complemented by next-generation sequencing techniques to efficiently obtain transcriptome-wide reactivity information (Choudhary et al., 2017; Kutchko and Laederach, 2017). This produces large datasets that demand for fast methods incorporating the probing data, which is met by our extension of IntaRNA introduced in the following.
For a given RNA–RNA interaction I (see Supplementary Material for detailed formalisms), its accessibility-based free energy is defined by . Therein, provides the hybridization energy from intermolecular base pairing while the terms represent the energy (penalty) needed to make the respective interacting subsequences unpaired/accessible (Mückstein et al., 2006). ED terms are defined by unpaired probabilities Prss of the subsequences via , where R is the gas constant and T the temperature. Detailed introductions on ED computation are provided e.g. in (Raden et al., 2018; Wright et al., 2018). Computation of unpaired probabilities can be guided by SHAPE data (Lorenz et al., 2016). While SHAPE-guided energy evaluations cannot be compared to unconstrained energy values (due to the introduced pseudo-energy terms), unpaired probabilities are compatible, since they are reflecting the accessible structure space rather than individual structures. Thus, SHAPE-constrained values can be directly used for ED and thus E computation while preserving comparability of the resulting energies.
Now, we show that SHAPE-guided accessibility prediction improves RNA–RNA interaction prediction. To this end, we study the probabilities of U1 small nuclear (sn)RNA interacting with pre-mRNA target sites, which is an established example of inter-molecular RNA interaction essential for RNA-splicing in eukaryotes. U1 is involved in pre-mRNA splicing by recognizing the 5’ site of introns via inter-molecular base pairing (Hertel and Graveley, 2005). Due to the dynamics and constraints imposed by the spliceosome, it is generally challenging to avoid false positive interaction predictions, which are either predictions of U1’s recognition site with random regions of the mRNA or predicted interactions of other U1 accessible regions with the mRNA. For that reason, we used U1 as an example to show that in vivo probing data effectively reduces false positive predictions in RNA–RNA interaction prediction.
SHAPE data for U1 was obtained from in vivo DMS-seq RNA structure probing of Arabidopsis thaliana (Ding et al., 2014). We selected the U1 homolog transcript bearing the largest secondary structure distance between the unconstrained and SHAPE-constrained structure prediction. The pre-mRNA sequences for five genes were extracted, which have been previously validated to perform U1-dependent splicing (Yeh et al., 2017). Figure 1a and b exemplify the effect of SHAPE-constrained predictions for ACT1 mRNA. Without SHAPE constraints, the splice site is predicted to interact with various regions of U1 with high probability; see Supplementary Material for formalisms. In contrast, the splice site’s interaction with U1’s recognition site is dominant with high specificity in SHAPE-guided IntaRNA predictions. Furthermore, SHAPE-guided prediction has a higher precision. Among all predicted interactions of U1 with the ACT1 mRNA, the ranking of the known interaction is improved from 9 to 3 in SHAPE-guided mode. When investigating the accessibility profile of U1 (Supplementary Material), this mainly results from an increased SHAPE-guided unpaired probability of U1’s recognition site and thus reduced ED penalties.
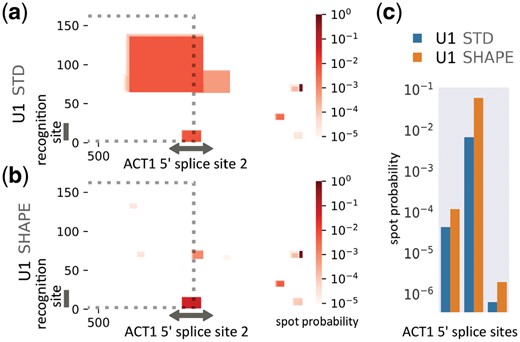
RNA–RNA interaction prediction between spliceosomal RNA U1 with ACT1 mRNA of A. thaliana. Interaction probabilities predicted between U1 (y-axis) and the region around the second intron splice site of ACT1 coding sequence mRNA using (a) unconstrained (STD) and (b) SHAPE-constrained accessibility estimates for U1. The dotted lines enclose U1 interactions with exon 2. (c) Spot probabilities of U1 recognition site (spot index = 8) interacting with the 5’ splice sites of ACT1 (spot = 1st intron index), with SHAPE constraints (orange) and without (blue) (Color version of this figure is available at Bioinformatics online.)
The interaction probability of U1’s recognition site with all three 5’ splice sites of ACT1’s coding sequence are increased when SHAPE data are incorporated (Fig. 1c). This effect results from a decreased number of false positive predictions (Fig. 1a and b). Following this trend, the probabilities of splice site recognition are improved among all the mRNAs and are on average 3.08 times higher (SHAPE/STD). Further details about data, evaluation and analyses are provided in the Supplementary Material.
Funding
This work was supported by Bundesministerium für Bildung und Forschung [031A538A RBC, 031L0106B] and Deutsche Forschungsgemeinschaft [BA 2168/14-1, BA 2168/16-1].
Conflict of Interest: none declared.
Acknowledgement
The authors thank Dr. Ronny Lorenz for discussions on SHAPE integration.
References


{kind=link}