





Abstract
The significance of long non-coding RNAs (lncRNAs) in many biological processes and diseases has gained intense interests over the past several years. However, computational identification of lncRNAs in a wide range of species remains challenging; it requires prior knowledge of well-established sequences and annotations or species-specific training data, but the reality is that only a limited number of species have high-quality sequences and annotations.
Here we first characterize lncRNAs in contrast to protein-coding RNAs based on feature relationship and find that the feature relationship between open reading frame length and guanine-cytosine (GC) content presents universally substantial divergence in lncRNAs and protein-coding RNAs, as observed in a broad variety of species. Based on the feature relationship, accordingly, we further present LGC, a novel algorithm for identifying lncRNAs that is able to accurately distinguish lncRNAs from protein-coding RNAs in a cross-species manner without any prior knowledge. As validated on large-scale empirical datasets, comparative results show that LGC outperforms existing algorithms by achieving higher accuracy, well-balanced sensitivity and specificity, and is robustly effective (>90% accuracy) in discriminating lncRNAs from protein-coding RNAs across diverse species that range from plants to mammals. To our knowledge, this study, for the first time, differentially characterizes lncRNAs and protein-coding RNAs based on feature relationship, which is further applied in computational identification of lncRNAs. Taken together, our study represents a significant advance in characterization and identification of lncRNAs and LGC thus bears broad potential utility for computational analysis of lncRNAs in a wide range of species.
LGC web server is publicly available at http://bigd.big.ac.cn/lgc/calculator. The scripts and data can be downloaded at http://bigd.big.ac.cn/biocode/tools/BT000004.
Supplementary data are available at Bioinformatics online.
1 Introduction
Long non-coding RNAs (lncRNAs) are prevalently expressed in a large number of organisms (Carninci et al., 2005; Djebali et al., 2012; Kapranov et al., 2007; Liu et al., 2015; Pennisi, 2010). Evidence has accumulated that lncRNAs play vital roles in biological processes including transcriptional regulation, post-transcriptional interference, translational control (Chen et al., 2017; Mercer et al., 2009; Quek et al., 2015; Rinn and Chang, 2012; Wilusz et al., 2009) and are implicated in the development of a variety of human diseases (Alam et al., 2017; Chen et al., 2013; Fang and Fullwood, 2016; Ma et al., 2015; Salhi et al., 2017). Although the rapid advancement in DNA sequencing technologies has led to an exponential increase in the number of lncRNAs (Iyer et al., 2015; Ma et al., 2015; Volders et al., 2015; Zhao et al., 2016), lncRNAs are often tissue/cell-specific (Alam et al., 2017; Cabili et al., 2011; Derrien et al., 2012) and lineage/species-specific (Derrien et al., 2012; Paralkar et al., 2014; Zheng et al., 2016) and thus a large number of novel lncRNAs are yet to be discovered. Experimental approaches (such as ribosome profiling and mass spectrometry) for coding potential detection could provide the most direct evidence but are very time-consuming and expensive yet with limited throughput. Therefore, computational approaches are in great demand for better characterizing the landscape of lncRNAs and identifying lncRNAs in a wide variety of species.
Over the past few years, several computational algorithms have been proposed to identify lncRNAs, which fall roughly into two classes: alignment-based algorithms (Achawanantakun et al., 2015; Hu et al., 2017; Kong et al., 2007; Lin et al., 2011; Liu et al., 2006; Sun et al., 2013a; Washietl et al., 2011) and alignment-free algorithms (Li et al., 2014; Sun et al., 2013b; Wang et al., 2013). Representative alignment-based algorithms include CPC (Coding Potential Calculator) (Kong et al., 2007), PhyloCSF (Phylogenetic Codon Substitution Frequencies) (Lin et al., 2011) and COME (coding potential calculation tool based on multiple features) (Hu et al., 2017). To distinguish lncRNAs from protein-coding transcripts, specifically, CPC uses sequence alignments against known proteins (Kong et al., 2007), PhyloCSF relies on multiple alignments of sequences from closely related species (Lin et al., 2011) and COME integrates multiple sequence-derived and experiment-based features (including DNA conservation, protein conservation, RNA structure conservation, guanine-cytosine (GC) content, expression, histone methylation) (Hu et al., 2017). Clearly, alignment-based algorithms are limited by the completeness of known proteins and the accuracy of DNA alignments and some of them are highly dependent on experiment-based features. Most importantly, they are incapable of identifying lncRNAs that are lineage/species-specific and become unreliable when no high-quality genome annotation is available. Additionally, alignment-based algorithms require prior sequence alignments and thus are exceedingly time-consuming, especially when more and more known sequences become available.
In contrast, alignment-free algorithms do not need any alignment but require high-quality protein-coding RNAs and lncRNAs as training data (Alam et al., 2014; Li et al., 2014; Sun et al., 2013b; Wang et al., 2013). Representative algorithms include CPAT (Coding Potential Assessment Tool) (Wang et al., 2013), CNCI (Coding-Non-Coding Index) (Sun et al., 2013b) and PLEK (predictor of lncRNAs and messenger RNAs based on an improved k-mer scheme) (Li et al., 2014). However, most of these algorithms are species-specific. These algorithms become unreliable when they are trained on data from one species and applied to data from another species (Achawanantakun et al., 2015; Li et al., 2014; Sun et al., 2013b). Moreover, alignment-free algorithms are heavily dependent on high-quality training data, but in reality, many species have low-quality or even no annotations, especially for newly sequenced species. It is reported that there are ∼8.7 million eukaryotic species on Earth and ∼90% species’ genomes are still waiting to be deciphered (Mora et al., 2011). Therefore, it is desirable to develop a more robust and effective algorithm that is able to accurately distinguish lncRNAs from protein-coding RNAs without the need of any prior information on alignment or training.
It would be straightforward to classify lncRNAs and protein-coding RNAs by taking account of sequence features. Although sequence features have already been factored in existing algorithms, for instance, ORF (open reading frame) length and coverage (Achawanantakun et al., 2015; Kong et al., 2007; Liu et al., 2006; Sun et al., 2013a; Wang et al., 2013), sequence similarity and conservation (Achawanantakun et al., 2015; Kong et al., 2007; Lin et al., 2011; Liu et al., 2006; Sun et al., 2013a; Washietl et al., 2011), nucleotide composition and codon usage (Achawanantakun et al., 2015; Hu et al., 2017; Li et al., 2014; Liu et al., 2006; Sun et al., 2013b; Wang et al., 2013), existing algorithms regard sequence features as independent variables and do not consider their potential biological relationship. Here we characterize lncRNAs in contrast to protein-coding RNAs based on a feature relationship between ORF length and GC content. As this feature relationship presents universally substantial divergence between lncRNAs and protein-coding RNAs as observed in a wide variety of species, we further propose LGC (ORF Length and GC content), a novel algorithm for robust and effective discrimination of lncRNAs from protein-coding RNAs. As testified on large-scale empirical datasets, LGC represents a significant advance over existing algorithms by identifying lncRNAs in a wide range of species not only effectively but also robustly. To our knowledge, this is the first to differentially characterize lncRNAs and protein-coding RNAs based on feature relationship, which is further applicable and effective in accurate identification of lncRNAs.
2 Materials and methods
2.1 Modelling the relationship between ORF length and GC content
To investigate the relationship between ORF length and GC content, we choose the top three longest ORFs (longer than 100 nt) for each sequence, as the transcribed ORFs are most likely from the top three. We divide ORFs into 100 groups based on their GC contents. Mean estimates of ORF length and GC content are used to estimate the parameters of Equation (3) by the least square method. Root mean square error (RMSE) is used as the criterion function for fitting the model of the expected ORF length from Equation (3) for both protein-coding RNAs and lncRNAs (Table 1).
Parameters for species-specific model
| Species | Protein-coding RNA | lncRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a 0 | a 1 | a 2 | a 3 | RMSEa | a 0 | a 1 | a 2 | a 3 | RMSEa | |
| Homo sapiens | 0.0247 | –0.1109 | 0.1781 | –0.0933 | 106.14 | 0.0018 | 0.0256 | –0.0577 | 0.0356 | 24.94 |
| Mus musculus | 0.0295 | –0.1241 | 0.1773 | –0.0776 | 109.30 | 0.0064 | –0.0035 | 0.0024 | –0.0043 | 21.36 |
| Danio rerio | 0.0499 | –0.2116 | 0.2772 | –0.0941 | 188.41 | 0.0200 | –0.0705 | 0.0919 | –0.0315 | 53.81 |
| Caenorhabditis elegans | 0.0707 | –0.4205 | 0.8338 | –0.5340 | 136.26 | 0.0067 | –0.0069 | 0.0084 | –0.0001 | 16.97 |
| Oryza sativa | 0.0246 | –0.1130 | 0.1803 | –0.0933 | 114.11 | 0.0177 | –0.0773 | 0.1568 | –0.1033 | 27.48 |
| Solanum lycopersicum | 0.0649 | –0.3437 | 0.5536 | –0.2249 | 177.46 | –0.0478 | 0.3571 | –0.7745 | 0.5419 | 66.07 |
| Species | Protein-coding RNA | lncRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a 0 | a 1 | a 2 | a 3 | RMSEa | a 0 | a 1 | a 2 | a 3 | RMSEa | |
| Homo sapiens | 0.0247 | –0.1109 | 0.1781 | –0.0933 | 106.14 | 0.0018 | 0.0256 | –0.0577 | 0.0356 | 24.94 |
| Mus musculus | 0.0295 | –0.1241 | 0.1773 | –0.0776 | 109.30 | 0.0064 | –0.0035 | 0.0024 | –0.0043 | 21.36 |
| Danio rerio | 0.0499 | –0.2116 | 0.2772 | –0.0941 | 188.41 | 0.0200 | –0.0705 | 0.0919 | –0.0315 | 53.81 |
| Caenorhabditis elegans | 0.0707 | –0.4205 | 0.8338 | –0.5340 | 136.26 | 0.0067 | –0.0069 | 0.0084 | –0.0001 | 16.97 |
| Oryza sativa | 0.0246 | –0.1130 | 0.1803 | –0.0933 | 114.11 | 0.0177 | –0.0773 | 0.1568 | –0.1033 | 27.48 |
| Solanum lycopersicum | 0.0649 | –0.3437 | 0.5536 | –0.2249 | 177.46 | –0.0478 | 0.3571 | –0.7745 | 0.5419 | 66.07 |
RMSE, root-mean-square error; see Equation (2) for more information on parameters (a0, a1, a2, a3).
Parameters for species-specific model
| Species | Protein-coding RNA | lncRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a 0 | a 1 | a 2 | a 3 | RMSEa | a 0 | a 1 | a 2 | a 3 | RMSEa | |
| Homo sapiens | 0.0247 | –0.1109 | 0.1781 | –0.0933 | 106.14 | 0.0018 | 0.0256 | –0.0577 | 0.0356 | 24.94 |
| Mus musculus | 0.0295 | –0.1241 | 0.1773 | –0.0776 | 109.30 | 0.0064 | –0.0035 | 0.0024 | –0.0043 | 21.36 |
| Danio rerio | 0.0499 | –0.2116 | 0.2772 | –0.0941 | 188.41 | 0.0200 | –0.0705 | 0.0919 | –0.0315 | 53.81 |
| Caenorhabditis elegans | 0.0707 | –0.4205 | 0.8338 | –0.5340 | 136.26 | 0.0067 | –0.0069 | 0.0084 | –0.0001 | 16.97 |
| Oryza sativa | 0.0246 | –0.1130 | 0.1803 | –0.0933 | 114.11 | 0.0177 | –0.0773 | 0.1568 | –0.1033 | 27.48 |
| Solanum lycopersicum | 0.0649 | –0.3437 | 0.5536 | –0.2249 | 177.46 | –0.0478 | 0.3571 | –0.7745 | 0.5419 | 66.07 |
| Species | Protein-coding RNA | lncRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a 0 | a 1 | a 2 | a 3 | RMSEa | a 0 | a 1 | a 2 | a 3 | RMSEa | |
| Homo sapiens | 0.0247 | –0.1109 | 0.1781 | –0.0933 | 106.14 | 0.0018 | 0.0256 | –0.0577 | 0.0356 | 24.94 |
| Mus musculus | 0.0295 | –0.1241 | 0.1773 | –0.0776 | 109.30 | 0.0064 | –0.0035 | 0.0024 | –0.0043 | 21.36 |
| Danio rerio | 0.0499 | –0.2116 | 0.2772 | –0.0941 | 188.41 | 0.0200 | –0.0705 | 0.0919 | –0.0315 | 53.81 |
| Caenorhabditis elegans | 0.0707 | –0.4205 | 0.8338 | –0.5340 | 136.26 | 0.0067 | –0.0069 | 0.0084 | –0.0001 | 16.97 |
| Oryza sativa | 0.0246 | –0.1130 | 0.1803 | –0.0933 | 114.11 | 0.0177 | –0.0773 | 0.1568 | –0.1033 | 27.48 |
| Solanum lycopersicum | 0.0649 | –0.3437 | 0.5536 | –0.2249 | 177.46 | –0.0478 | 0.3571 | –0.7745 | 0.5419 | 66.07 |
RMSE, root-mean-square error; see Equation (2) for more information on parameters (a0, a1, a2, a3).
2.2 Maximum likelihood estimation of coding potential
Symbols used in calculating coding potential score
| Symbol | Definition |
|---|---|
| PA | Probability of adenine |
| PT | Probability of thymine |
| PG | Probability of guanine |
| PC | Probability of cytosine |
| PGC | GC contenta |
| F | Stop-codon probability |
| f c | Stop-codon probability in coding sequence |
| f nc | Stop-codon probability in non-coding sequence |
| E(l) | Expected ORF lengtha |
| Pi | Frequency of ORFs with the length of i nt |
| p c | Probability of ORF in coding sequence |
| p nc | Probability of ORF in non-coding sequence |
| L | Coding potential score |
| Symbol | Definition |
|---|---|
| PA | Probability of adenine |
| PT | Probability of thymine |
| PG | Probability of guanine |
| PC | Probability of cytosine |
| PGC | GC contenta |
| F | Stop-codon probability |
| f c | Stop-codon probability in coding sequence |
| f nc | Stop-codon probability in non-coding sequence |
| E(l) | Expected ORF lengtha |
| Pi | Frequency of ORFs with the length of i nt |
| p c | Probability of ORF in coding sequence |
| p nc | Probability of ORF in non-coding sequence |
| L | Coding potential score |
PGC and E(l) were used to train the model of LGC.
Symbols used in calculating coding potential score
| Symbol | Definition |
|---|---|
| PA | Probability of adenine |
| PT | Probability of thymine |
| PG | Probability of guanine |
| PC | Probability of cytosine |
| PGC | GC contenta |
| F | Stop-codon probability |
| f c | Stop-codon probability in coding sequence |
| f nc | Stop-codon probability in non-coding sequence |
| E(l) | Expected ORF lengtha |
| Pi | Frequency of ORFs with the length of i nt |
| p c | Probability of ORF in coding sequence |
| p nc | Probability of ORF in non-coding sequence |
| L | Coding potential score |
| Symbol | Definition |
|---|---|
| PA | Probability of adenine |
| PT | Probability of thymine |
| PG | Probability of guanine |
| PC | Probability of cytosine |
| PGC | GC contenta |
| F | Stop-codon probability |
| f c | Stop-codon probability in coding sequence |
| f nc | Stop-codon probability in non-coding sequence |
| E(l) | Expected ORF lengtha |
| Pi | Frequency of ORFs with the length of i nt |
| p c | Probability of ORF in coding sequence |
| p nc | Probability of ORF in non-coding sequence |
| L | Coding potential score |
PGC and E(l) were used to train the model of LGC.
2.3 Performance evaluation of LGC
Protein-coding RNAs (38 811 transcripts) and lncRNAs (27 669 transcripts) of human (Supplementary Table S1) are used to build LGC. Ten-fold cross-validation shows that LGC achieves very high accuracy on human data, with an AUC of 0.981 (Supplementary Fig. S2). LGC is evaluated by comparison with several existing popular algorithms, including CPC (Kong et al., 2007), CPAT (Wang et al., 2013), CNCI (Sun et al., 2013b) and PLEK (Li et al., 2014). LGC, CPC and PLEK can be used in a cross-species manner that do not require any training or specific model. CNCI is also used in a cross-species manner, but uses two specific models, namely, ‘ve’ and ‘pl’, to identify lncRNAs in animals (human, mouse, zebrafish and worm) and plants (rice and tomato), respectively. Contrastingly, CPAT uses species-specific training data to build specific models. Specifically, it adopts species-specific logistic regression models to calculate coding probability and sets different cut-offs, viz., 0.36 for human, 0.44 for mouse, 0.38 for zebrafish and 0.39 for worm. Due to the lack of a prebuilt model for plant, the logistic regression model of human is additionally applied to rice and tomato during performance comparison. We compare LGC with algorithms that can be used in a cross-species manner or adopt specific models. All datasets used for comparisons are summarized in Supplementary Table S1. To reduce any bias from unequal sampling size of lncRNAs and protein-coding RNAs, we randomly select protein-coding RNAs with the equal number of lncRNAs.
2.4 Data collection
A total of six representative organisms are used in this study, including two mammals (human and mouse), one vertebrate (zebrafish), one invertebrate (worm) and two plants (rice and tomato). Protein-coding RNAs for human and mouse are both collected from NCBI RefSeq (Pruitt et al., 2007) and their corresponding lncRNAs are obtained from GENCODE version 22 (Harrow et al., 2012) and GENCODE version M7 (Mudge and Harrow, 2015), respectively. For the remaining organisms, both protein-coding RNAs and ncRNAs are downloaded from Ensembl (Cunningham et al., 2015). To obtain lncRNAs, ncRNAs < 200 nt are excluded. All detailed information of these datasets is summarized in Supplementary Table S1.
Also, to examine the robustness of LGC for a wider diversity of species that range from plants to mammals, we set up a more comprehensive dataset by collecting all curated protein-coding RNAs (accession prefixed with NM) and ncRNAs (accession prefixed with NR) from NCBI RefSeq (Pruitt et al., 2007).
2.5 Availability
The package of LGC can be downloaded for academic use only at BioCode (a source code archive for bioinformatics software tools; http://bigd.big.ac.cn/biocode) in the BIG Data Center (2018), with accession number BT000004. In addition, a web server is publicly available at http://bigd.big.ac.cn/lgc/calculator.
3 Results and discussion
3.1 Characterization of protein-coding RNAs and lncRNAs based on feature relationship
It is extensively documented that in protein-coding sequences ORF length is dominantly determined by GC content, since base composition of translational stop codons (TAG, TAA and TGA) is biased toward low GC content (Oliver and Marín, 1996; Xia et al., 2003, 2006). If a sequence is AT-rich, it is most likely that stop codons would appear earlier, resulting in a shorter ORF; conversely, a GC-rich sequence tends to have longer ORF because it is less likely to have stop codons earlier (Oliver and Marín, 1996). Considering that protein-coding RNAs differ from lncRNAs in possessing significantly longer ORFs (Achawanantakun et al., 2015; Kong et al., 2007; Sun et al., 2013a; Wang et al., 2013), it is possible that protein-coding RNAs and lncRNAs may present different relationships between GC content and ORF length. Of course, ORF length, as one of the important features, has been widely used by the existing algorithms in coding potential prediction (Achawanantakun et al., 2015; Kong et al., 2007; Liu et al., 2006; Sun et al., 2013a; Wang et al., 2013). However, the two features—ORF length and GC content—are often regarded as independent, and their relationship has not been well characterized in protein-coding RNAs and lncRNAs. Therefore, we model the relationship between ORF length and GC content and hypothesize that this relationship can be used to differentially characterize protein-coding RNAs and lncRNAs. In addition to ORF length and GC content, undoubtedly, it cannot rule out the possibility that other features [such as, codon usage bias and gene length (Eyre-Walker, 1996) codon usage bias and GC content (Novembre, 2002; Plotkin and Kudla, 2011)] may present a similar relationship that can be used for lncRNA identification.
To test the hypothesis, we collect protein-coding RNAs and lncRNAs from six representative organisms (Supplementary Table S1) and examine their corresponding relationships between ORF length and GC content (based on Equation 3; see Section 2). Consistent with our expectations, protein-coding RNAs and lncRNAs present strikingly different relationships in all investigated organisms (Fig. 1). An obvious inverted V-shape curve is observed in protein-coding RNAs, i.e. ORF length increases with GC content for low-GC genes, while decreases for high-GC genes. This is well consistent with pervious findings that selection against cytosine usage (prone to mutation to T/U; e.g. CAR to TAR and CGA to TGA) (Xia et al., 2006) in GC-rich genes may contribute to negative correlation between GC content and ORF length. When compared with protein-coding RNAs, contrastingly, curves are extremely flat in lncRNAs. Overall, these results show that protein-coding RNAs and lncRNAs exhibit significant and universal heterogeneity in the relationship between ORF Length and GC content (LGC model). Thus, based on the LGC model, we further explore whether such heterogeneity can be used to effectively distinguish lncRNAs from protein-coding RNAs for a wide variety of species.
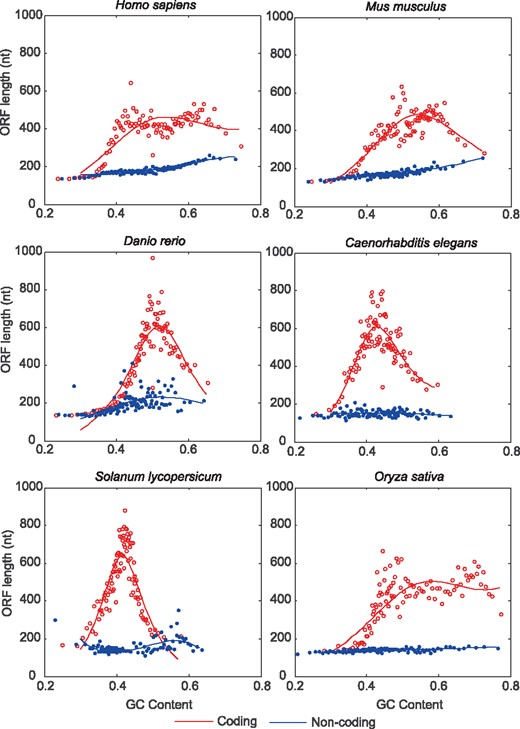
Relationship between ORF length and GC content for protein-coding RNAs (red circles) and lncRNAs (blue dots), respectively. For each transcript, the top three longest ORFs (longer than 100 nt) are used. ORFs are grouped into 100 bins based on their GC contents and each dot represents the average estimate for each bin (Color version of this figure is available at Bioinformatics online.)
3.2 Application of the LGC model in lncRNA identification
To apply the LGC model in the identification of lncRNAs, we first estimate parameters in Equations (2) and (3) (see Section 2) using all lncRNAs and protein-coding RNAs for each species and build species-specific LGC (Table 1). We then employ these parameters’ estimates to calculate the coding potential score (Equation 4), which is an indicator to distinguish lncRNAs from protein-coding RNAs. As validated on empirical datasets from six representative species (Table 3), we find that species-specific LGC model achieves high accuracy (>0.88) in each species and performs well in the identification of both protein-coding RNAs and lncRNAs as indicated by well-balanced sensitivities and specificities in most datasets. These results suggest that the LGC model is indeed applicable for identifying lncRNAs in a wide range of species.
Performance of LGC based on species-specific model and human model
| Species | Species-specific model | Human model | ||||
|---|---|---|---|---|---|---|
| Acc | Sen | Spe | Acc | Sen | Spe | |
| H.sapiens | 0.945 | 0.964 | 0.925 | 0.945 | 0.964 | 0.925 |
| M.musculus | 0.936 | 0.948 | 0.924 | 0.938 | 0.960 | 0.916 |
| D.rerio | 0.884 | 0.881 | 0.906 | 0.920 | 0.945 | 0.895 |
| C.elegans | 0.933 | 0.870 | 0.996 | 0.946 | 0.900 | 0.991 |
| S.lycopersicum | 0.887 | 0.778 | 0.995 | 0.907 | 0.818 | 0.996 |
| O.sativa | 0.963 | 0.927 | 0.999 | 0.961 | 0.923 | 0.999 |
| Species | Species-specific model | Human model | ||||
|---|---|---|---|---|---|---|
| Acc | Sen | Spe | Acc | Sen | Spe | |
| H.sapiens | 0.945 | 0.964 | 0.925 | 0.945 | 0.964 | 0.925 |
| M.musculus | 0.936 | 0.948 | 0.924 | 0.938 | 0.960 | 0.916 |
| D.rerio | 0.884 | 0.881 | 0.906 | 0.920 | 0.945 | 0.895 |
| C.elegans | 0.933 | 0.870 | 0.996 | 0.946 | 0.900 | 0.991 |
| S.lycopersicum | 0.887 | 0.778 | 0.995 | 0.907 | 0.818 | 0.996 |
| O.sativa | 0.963 | 0.927 | 0.999 | 0.961 | 0.923 | 0.999 |
Note: The species-specific model indicates that LGC is built based on species-specific protein-coding RNAs and lncRNAs. The human model indicates that LGC is built based only on human protein-coding RNAs and lncRNAs. These models are compared in terms of accuracy, sensitivity and specificity, where numbers in bold represent the better performance.
Acc, accuracy; Sen, sensitivity; Spe, specificity.
Performance of LGC based on species-specific model and human model
| Species | Species-specific model | Human model | ||||
|---|---|---|---|---|---|---|
| Acc | Sen | Spe | Acc | Sen | Spe | |
| H.sapiens | 0.945 | 0.964 | 0.925 | 0.945 | 0.964 | 0.925 |
| M.musculus | 0.936 | 0.948 | 0.924 | 0.938 | 0.960 | 0.916 |
| D.rerio | 0.884 | 0.881 | 0.906 | 0.920 | 0.945 | 0.895 |
| C.elegans | 0.933 | 0.870 | 0.996 | 0.946 | 0.900 | 0.991 |
| S.lycopersicum | 0.887 | 0.778 | 0.995 | 0.907 | 0.818 | 0.996 |
| O.sativa | 0.963 | 0.927 | 0.999 | 0.961 | 0.923 | 0.999 |
| Species | Species-specific model | Human model | ||||
|---|---|---|---|---|---|---|
| Acc | Sen | Spe | Acc | Sen | Spe | |
| H.sapiens | 0.945 | 0.964 | 0.925 | 0.945 | 0.964 | 0.925 |
| M.musculus | 0.936 | 0.948 | 0.924 | 0.938 | 0.960 | 0.916 |
| D.rerio | 0.884 | 0.881 | 0.906 | 0.920 | 0.945 | 0.895 |
| C.elegans | 0.933 | 0.870 | 0.996 | 0.946 | 0.900 | 0.991 |
| S.lycopersicum | 0.887 | 0.778 | 0.995 | 0.907 | 0.818 | 0.996 |
| O.sativa | 0.963 | 0.927 | 0.999 | 0.961 | 0.923 | 0.999 |
Note: The species-specific model indicates that LGC is built based on species-specific protein-coding RNAs and lncRNAs. The human model indicates that LGC is built based only on human protein-coding RNAs and lncRNAs. These models are compared in terms of accuracy, sensitivity and specificity, where numbers in bold represent the better performance.
Acc, accuracy; Sen, sensitivity; Spe, specificity.
To further test the universality of the LGC model across different species, we compare the performances of species-specific LGC model against the LGC model built based merely on human data (whose quality is believed to be relatively higher). Strikingly, the LGC model based on human data overall shows better performances than the species-specific LGC models in terms of accuracy, specificity and sensitivity (Table 3); it achieves higher accuracies (>0.9) in all six organisms, with both sensitivities and specificities >0.9 in most datasets. Although the LGC model is built based on human data, high accuracy is achieved not only for mammals and vertebrates but also for invertebrates and plants. This is most likely caused by both larger-quantity and higher-quality of human data. Accordingly, the LGC model built by human data is superior to that based on species-specific data, as testified on multiple empirical real datasets (Table 3). These results suggest that the LGC model is universally applicable, guaranteeing that the LGC model can be used in a cross-species manner without requiring species-specific data.
3.3 Effective discrimination of lncRNAs from protein-coding RNAs
To test the effectiveness of the LGC model and to examine its performance in discriminating lncRNAs from protein-coding RNAs, we further evaluate it on data from the six representative organisms by comparison with several popular algorithms including CPC (Kong et al., 2007), CNCI (Sun et al., 2013b) and PLEK (Li et al., 2014) that can be used in a cross-species manner (see Section 2). In what follows, the LGC model built on human data (as detailed earlier), is used in all comparisons.
Comparative evaluations regarding accuracy, specificity and sensitivity show that LGC outperforms existing algorithms, significantly and robustly across different species (Fig. 2). Specifically, LGC overall achieves higher accuracies for all six organisms (>0.9); it outperforms PLEK in non-human species, CNCI in non-mammal species, and CPC in human, mouse, zebrafish and rice. Considering the average accuracy over all six species (Table 4), LGC obtains the highest average accuracy (0.936) compared with CPC (0.906), CNCI (0.896) and PLEK (0.848). Moreover, LGC yields better average specificity of 0.954 across all six species than the other algorithms (Table 4); it outperforms CNCI and PLEK in zebrafish and rice, and CPC in human, mouse, zebrafish and rice (and performs comparably in the remaining cases) (Fig. 2). Regarding sensitivity, LGC achieves the average sensitivity at 0.918 (just follows CPC at 0.994), better than CNCI at 0.850, and PLEK at 0.760 (Table 4); it outperforms CNCI in human, mouse, worm and tomato, and PLEK in non-human species (Fig. 2).
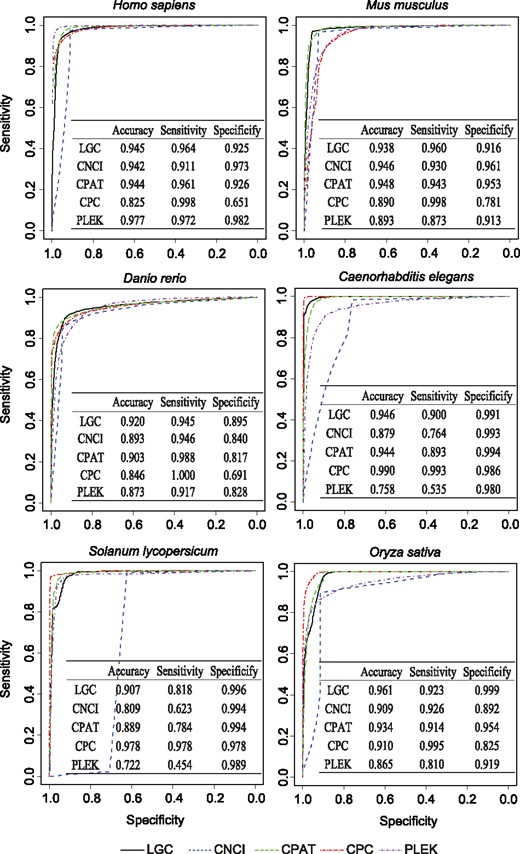
Performances of LGC, CNCI, CPAT, CPC and PLEK. LGC, CPC, CNCI and PLEK can be used in a cross-species manner, while CPAT uses specific models and cut-offs for different species (see Section 2)
Estimates of accuracy, sensitivity and specificity averaged over six representative organisms
| Algorithm | LGC | CNCI | CPAT | CPC | PLEK |
|---|---|---|---|---|---|
| Accuracy | 0.936 | 0.896 | 0.927 | 0.906 | 0.848 |
| Sensitivity | 0.918 | 0.850 | 0.914 | 0.994 | 0.760 |
| Specificity | 0.954 | 0.942 | 0.940 | 0.819 | 0.935 |
| Algorithm | LGC | CNCI | CPAT | CPC | PLEK |
|---|---|---|---|---|---|
| Accuracy | 0.936 | 0.896 | 0.927 | 0.906 | 0.848 |
| Sensitivity | 0.918 | 0.850 | 0.914 | 0.994 | 0.760 |
| Specificity | 0.954 | 0.942 | 0.940 | 0.819 | 0.935 |
Note: Numbers in bold represent the better performance.
Estimates of accuracy, sensitivity and specificity averaged over six representative organisms
| Algorithm | LGC | CNCI | CPAT | CPC | PLEK |
|---|---|---|---|---|---|
| Accuracy | 0.936 | 0.896 | 0.927 | 0.906 | 0.848 |
| Sensitivity | 0.918 | 0.850 | 0.914 | 0.994 | 0.760 |
| Specificity | 0.954 | 0.942 | 0.940 | 0.819 | 0.935 |
| Algorithm | LGC | CNCI | CPAT | CPC | PLEK |
|---|---|---|---|---|---|
| Accuracy | 0.936 | 0.896 | 0.927 | 0.906 | 0.848 |
| Sensitivity | 0.918 | 0.850 | 0.914 | 0.994 | 0.760 |
| Specificity | 0.954 | 0.942 | 0.940 | 0.819 | 0.935 |
Note: Numbers in bold represent the better performance.
Strikingly, LGC provides well-balanced sensitivity and specificity (both higher than 82%), which is consistently observed for all examined species (Fig. 2). Contrary to this, existing algorithms show poor balance between sensitivity and specificity; CPC yields extremely unbalanced sensitivity and specificity in human, mouse and zebrafish (for instance, 0.998 and 0.651 in human, respectively), CNCI presents unbalanced sensitivity and specificity in worm and tomato (for instance, 0.764 and 0.993 in worm, respectively) (consistent with the previous study in Sun et al., 2013b), and PLEK exhibits unbalanced sensitivity and specificity in worm and tomato (for instance, 0.535 and 0.980 in worm, respectively). Taken together, these results clearly show that LGC achieves a good balance between sensitivity and specificity and is capable of discriminating lncRNAs from protein-coding RNAs more accurately than the existing algorithms.
To further evaluate the performance of LGC, we also compare it with CPAT (Wang et al., 2013), which requires appropriate training to build specific models with different cut-off values (see details in Section 2). Albeit CPAT uses species-specific models, we find that LGC overall performs better than CPAT (Fig. 2 and Table 4). Specifically, it performs comparably with CPAT in human, mouse, and worm (with the accuracy around 0.94), and outperforms CPAT in zebrafish, tomato, and rice (Fig. 2). Although CPAT builds species-specific models for human, mouse, zebrafish and fly, it does not perform well as expected. In zebrafish, CPAT shows poor balance between sensitivity (0.988) and specificity (0.817), whereas LGC yields sensitivity at 0.945 and specificity at 0.895. This may because that training data of different models show unequal qualities, and the robust performance of human, mouse and fly models of CPAT are attributable to the high quality of training datasets. Also, it is noted that species-specific algorithms have significant limitation in application. As no prebuilt models are available for plants, we apply human model of CPAT to tomato and rice. However, the human model of CPAT presents unbalanced sensitivity (at 0.784) and specificity (at 0.994) in tomato, whereas LGC yields sensitivity at 0.818 and specificity at 0.996. Given that CPAT is heavily dependent on high-quality training data and many species presently may still have low-quality or even no training data, LGC bears broad utility for computational analysis of lncRNAs in a wide range of species.
3.4 Robustness in a wide diversity of species
To further examine the robustness of LGC for a wider diversity of species, we set up a more comprehensive dataset by collecting all curated protein-coding RNAs (accession prefixed with NM) and ncRNAs (accession prefixed with NR) from NCBI RefSeq (Pruitt et al., 2007). All protein-coding RNAs and ncRNAs are classified into: mammals (127 903 protein-coding RNAs from 81 species and 23 644 ncRNAs from 26 species), vertebrates (53 239 protein-coding RNAs from 59 species and 2582 ncRNAs from 9 species), invertebrates (68 229 protein-coding RNAs from 42 species and 29 527 ncRNAs from 11 species) and plants (97 119 protein-coding RNAs from 34 species and 1795 ncRNAs from 10 species).
We test the performance of LGC on this more comprehensive dataset derived from a larger number of species and compare it against existing algorithms that can be used in a cross-species manner without requiring any species-specific training or model. Accordingly, only PLEK, albeit built on human data, can be used for a wide range of species (Li et al., 2014), whereas other algorithms are unsuitable for this comparison [as CNCI is limited to two specific models, namely, ‘ve’ for vertebrates, and ‘pl’ for plants (Sun et al., 2013b), CPC depends on sequence alignments against known proteins (Kong et al., 2007), which are completely identical to the dataset obtained from NCBI RefSeq (Pruitt et al., 2007)]. Comparative results show that in general LGC performs more stable and achieves higher accuracy (>0.9 for most datasets) in the identification of both protein-coding and ncRNAs (Fig. 3). In contrast, PLEK, based on a k-mer scheme and a support vector machine algorithm (Li et al., 2014), performs poorly and shows an obvious imbalance in its ability to identify both protein-coding and non-coding RNAs for all investigated cases (Fig. 3). In addition, PLEK presents unstable varied performances among species within groups of plants, invertebrates, vertebrates and mammal, whereas LGC achieves robust higher accuracies in almost all datasets (Fig. 3, Supplementary Tables S2 and S3). Collectively, these results indicate that LGC is robust in accurately discriminating lncRNAs from protein-coding RNAs in a wide variety of species.
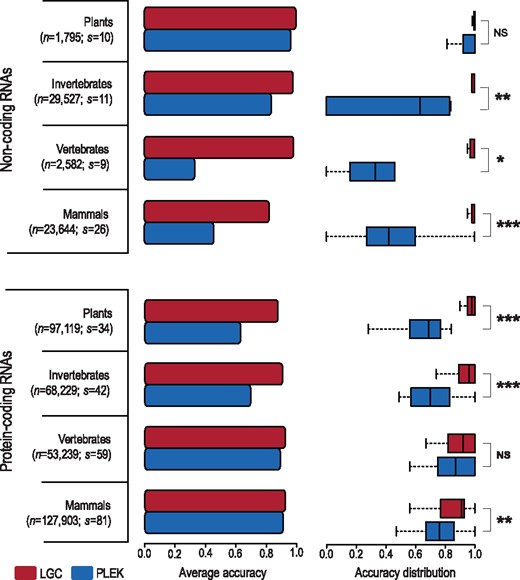
Accuracy of LGC (upper bar) and PLEK (lower bar) on protein-coding RNAs and non-coding RNAs from NCBI RefSeq. The number of sequences (n) as well as the number of species (s) is labelled. The boxes depict data between the 25th and 75th percentiles with central horizontal lines representing the median values. The Wilcoxon test is used to evaluate the significance level when comparing the accuracy between LGC and PLEK, and P-value is indicated by NS (Not Significant) > 0.05, ‘*’ < 0.05, ‘**’ < 10−3 and ‘***’ < 10−5. Comparison results for each species are listed in Supplementary Table S2 (non-coding RNA) and Supplementary Table S3 (protein-coding RNA)
4 Conclusion
To our knowledge, our study is the first to differentially characterize lncRNAs and protein-coding RNAs based on a feature relationship between ORF length and GC content, on the grounds that lncRNAs and protein-coding RNAs present considerable divergence in terms of this relationship, which is consistently and universally detected in a wide range of species. Hence, we further present LGC, a novel algorithm to discriminate lncRNAs from protein-coding RNAs based on this feature relationship. As demonstrated in multiple empirical datasets across a wide diversity of species, LGC is superior to existing algorithms by achieving higher accuracy and well-balanced sensitivity and specificity. In addition, LGC is able to accurately and robustly distinguish lncRNAs from protein-coding RNAs in a cross-species manner without the need for species-specific adjustments. Overall, LGC represents a simple, robust and powerful algorithm for characterization and identification of lncRNAs in a wide range of species, providing a significant advance for computational analysis of lncRNAs.
Acknowledgements
We thank Shuhui Song, Lili Hao and Shixiang Sun for valuable comments on this work.
Funding
This work was supported by Strategic Priority Research Programme of the Chinese Academy of Sciences [XDB13040500 and XDA08020102 to Z.Z.]; National Key Research and Development Programme of China [2017YFC0907502 and 2015AA020108 to Z.Z.; 2016YFE0206600 to Y.B.]; International Partnership Programme of the Chinese Academy of Sciences [153F11KYSB20160008]; National Natural Science Foundation of China [31200978 to L.M.]; The 100-Talent Programme of Chinese Academy of Sciences to Z.Z. and Y.B.; The Open Biodiversity and Health Big Data Initiative of IUBS [Y.B.]; The 13th Five-year Informatization Plan of Chinese Academy of Sciences [XXH13505-05 to Y.B.]; The King Abdullah University of Science and Technology (KAUST) Base Research Funds [BAS/1/1606-01-01 to VBB]. Funding for open access charge: Strategic Priority Research Programme of the Chinese Academy of Sciences.
Conflict of Interest: none declared.
References
BIG Data Center Members. (


{kind=link}
{kind=link}
{kind=link}