





Abstract
MicroRNA (miRNA) and alternative splicing (AS)-mediated post-transcriptional regulation has been extensively studied in most eukaryotes. However, the interplay between AS and miRNAs has not been explored in plants. To our knowledge, the overall profile of miRNA target sites in circular RNAs (circRNA) generated by alternative back splicing has never been reported previously. To address the challenge, we identified miRNA target sites located in alternatively spliced regions of the linear and circular splice isoforms using the up-to-date single-molecule real-time (SMRT) isoform sequencing (Iso-Seq) and Illumina sequencing data in eleven plant species.
In total, we identified 399 401 and 114 574 AS events from linear and circular RNAs, respectively. Among them, there were 64 781 and 41 146 miRNA target sites located in linear and circular AS region, respectively. In addition, we found 38 913 circRNAs to be overlapping with 45 648 AS events of its own parent isoforms, suggesting circRNA regulation of AS of linear RNAs by forming R-loop with the genomic locus. Here, we present a comprehensive database of miRNA targets in alternatively spliced linear and circRNAs (ASmiR) and a web server for deposition and identification of miRNA target sites located in the alternatively spliced region of linear and circular RNAs. This database is accompanied by an easy-to-use web query interface for meaningful downstream analysis. Plant research community can submit user-defined datasets to the web service to search AS regions harboring small RNA target sites. In conclusion, this study provides an unprecedented resource to understand regulatory relationships between miRNAs and AS in both gymnosperms and angiosperms.
The readily accessible database and web-based tools are available at http://forestry.fafu.edu.cn/bioinfor/db/ASmiR.
Supplementary data are available at Bioinformatics online.
1 Introduction
Gene expression, a fundamental process critical for growth and development of all organisms and their response to environmental cues/stresses, is regulated at several levels. Multiple mechanisms including primary transcripts (pre-mRNA) splicing and microRNA (miRNA)-directed cleavage of transcripts regulate and fine-tune gene expression at the post-transcriptional level. MicroRNAs are short (20–24 nt) single-stranded RNAs that down-regulate gene expression by translation inhibition (Brodersen et al., 2008) or mRNA degradation (Henderson and Jacobsen, 2008). Because of the biological importance, miRNAs from 33 representative species across vascular plants have been investigated (Montes et al., 2014; Xia et al., 2015). For example, miRNA families, such as miR482/miR2118 from Picea abies, trigger phasiRNA by targeting nucleotide-binding leucine-rich repeat (NB-LRR) genes and non-coding PHAS loci (Xia et al., 2015). In Populus trichocarpa, miRNAs are associated with wood formation (Lu et al., 2005), stress-responses (Lu et al., 2008; Shuai et al., 2013) and the transition from juvenile to reproductive phase (Wang et al., 2011).
In eukaryotes, pre-mRNAs from genes containing several introns are spliced to generate mature mRNAs. Alternative splicing (AS) of pre-mRNAs, a process that generates multiple distinct transcripts from a single multi-exon gene (Reddy, 2007) is prevalent in plants, such as Arabidopsis thaliana (Filichkin et al., 2010), Oryza sativa (Lu et al., 2010; Zhang et al., 2010), Zea mays (Li et al., 2010), Sorghum bicolor (Abdel-Ghany et al., 2016), Phyllostachys edulis (Li et al., 2016a, b, c) and P.trichocarpa (Filichkin et al., 2018). Recent single-molecule real-time (SMRT) isoform sequencing (Iso-Seq) results also showed that the AS is widespread in Arabidopsis thaliana (Li et al., 2016a, b, c), Sorghum bicolor (Abdel-Ghany et al., 2016) and Zea mays (Wang et al., 2016) and Phyllostachys edulis (Wang et al., 2017). However, a database with all the up-to-date AS events from Iso-Seq is lacking. Although the two-conifer genomes of Picea abies (Nystedt et al., 2013) and Pinus taeda (Neale et al., 2014) have been sequenced, AS events have never been reported in ancient gymnosperms. Populus, Phyllostachys edulis and Eucalyptus grandis are three fast-growing woody perennials (Myburg et al., 2014; Peng et al., 2013; Tuskan et al., 2006). Eucalyptus, a highly adaptable plant, is the most widely planted tree, however, the AS of Eucalyptus has not been investigated (Myburg et al., 2014). Though the AS in Populus and Phyllostachys edulis has been reported (Baek et al., 2008; Bao et al., 2013; Filichkin et al., 2018; Li et al., 2016a, b, c; Srivastava et al., 2009; Tang et al., 2015; Wong et al., 2011; Zhao et al., 2014a,b), a database for deposition of these AS events in these plant species is still lacking. The lack of a comprehensive database of AS is hindering research progress in woody perennials.
Circular RNAs (circRNA) were initially found to exist using the electron microscope in 1979 (Hsu and Coca-Prados, 1979). With the advent of high throughput sequencing approaches, hundreds of thousands circular RNAs were identified in humans, animals and plants (Jeck et al., 2013; Lu et al., 2015; Memczak et al., 2013; Salzman et al., 2012; Ye et al., 2015). Studies in plants showed that naturally expressed circRNAs repress the biogenesis of miRNA by sequestering the dicing complex (Li et al., 2016a,b,c) or regulate exon skipping by binding to its cognate gene and causing transcriptional pause (Conn et al., 2017). In addition to AS from linear RNAs, alternative back-splicing that generates circRNA variants, which may have distinct functions, is also reported (Zhang et al., 2016a,b). Although several databases have been designed for circular RNA in plants, such as PlantCircNet (Zhang et al., 2017a,b), AtCircDB (Ye et al., 2017a, b) and PlantcircBase (Chu et al., 2017), a database for comprehensive analysis of circRNA variants produced by back-splicing is seldom reported.
At present, high-quality reference genomes for several model plants and fast-growing woody species are available, providing the necessary sequence information for studying both AS and miRNA. However, relatively little research has focused on the interplay between miRNA and AS. A previous study showed that miRNA binding sites in DCL1 and TAS genes are regulated by AS in Arabidopsis (Yang et al., 2012). However, the genome-wide interplay between miRNA and AS from linear and circular RNAs remains largely unknown. To address this need, we developed a comprehensive database called ASmiR to identify all splice isoforms from linear and circRNAs originated from the same locus with or without miRNA target sites. At the same time, we also provide a powerful web-based service to identify AS regions including the target sites of miRNAs in the user submitted datasets. ASmiR will be a unique resource to accelerate the research of post-transcriptional regulation in plants.
2 Materials and methods
2.1 Data source
Populus trichocarpa genome sequence and annotation (v3.0) were obtained from ftp sites of Phytozome v11.0 (https://phytozome.jgi.doe.gov). Genome and gene models of Phyllostachys edulis were downloaded from bambooGDB (Zhao et al., 2014a, b). Genome and gene models for Eucalyptus (v2.0) were obtained from ftp sites of Phytozome v10 (https://phytozome.jgi.doe.gov). Genome sequences and gene models for Picea abies (Nystedt et al., 2013) and Pinus taeda (Neale et al., 2014) were downloaded from http://congenie.org. The genome versions for Arabidopsis thaliana, Oryza sativa, Glycine max, Solanum lycopersicum, Sorghum bicolor and Zea mays were TAIR10, TIGR7, Glycine_max.V1.0.31, S_lycopersicum.2.50, Sorghum_bicolor.Sorbi1.31 and Zea_mays.AGPv4, respectively. Gene ontology (GO) annotations in eleven different species were obtained using BLAST2GO (Conesa et al., 2005). Orthologous genes were identified using InParanoid algorithms to provide a comparison among different species (Ostlund et al., 2010).
SMRT Iso-Seq reads were obtained from Arabidopsis thaliana (Li et al., 2016a, b, c), Zea mays (Wang et al., 2016), Sorghum bicolor (Abdel-Ghany et al., 2016) and Phyllostachys edulis (Wang et al., 2017), respectively. RNA-Seq libraries were collected from Populus (Geraldes et al., 2011; Liang et al., 2014), Phyllostachys edulis (Peng et al., 2013), Eucalyptus (Mangwanda et al., 2015; Oates et al., 2015), Picea abies (Nystedt et al., 2013) and Pinus taeda (Neale et al., 2014), respectively. In total, 312 RNA-Seq libraries were downloaded from NCBI's Sequence Read Archive (SRA) for Arabidopsis thaliana (Hernando et al., 2015), Oryza sativa (Oono et al., 2013; Zhai et al., 2013; Zhang et al., 2010), Glycine max (Shen et al., 2014a, b), Solanum lycopersicum (Consortium, 2012; Richard et al., 2015; Zhang et al., 2016a, b) and Sorghum bicolor (Makita et al., 2015), respectively. In addition, miRNA sequences of P.trichocarpa, Eucalyptus, Picea abies, Pinus taeda, Arabidopsis thaliana, Oryza sativa, Glycine max, Solanum lycopersicum, Sorghum bicolor and Zea mays were downloaded from miRBase 21 (Griffiths-Jones et al., 2008).
2.2 Identification of AS, miRNA target sites and the interplay
The bioinformatics workflow for detecting miRNA target sites in alternatively spliced regions (ASR) is illustrated in Figure 1A. Firstly, RNA-Seq dataset from SRA was transferred into FASTQ with NCBI’s SRA Toolkit (Leinonen et al., 2011). Then RNA-Seq reads were aligned against corresponding genome sequences using TopHat-2.0.11 with an anchor length of more than 8 nt for spliced alignments (Langmead et al., 2009). The mapped reads were assembled with Cufflinks v2.1.1 using following parameters: -F 0.05 –A 0.01 –I 100000 –min-intron-length 30 (Trapnell et al., 2012). The transcripts from genome-guided assembly were used to infer AS events with rMATS.3.2.2 using default parameters (Shen et al., 2014a, b).
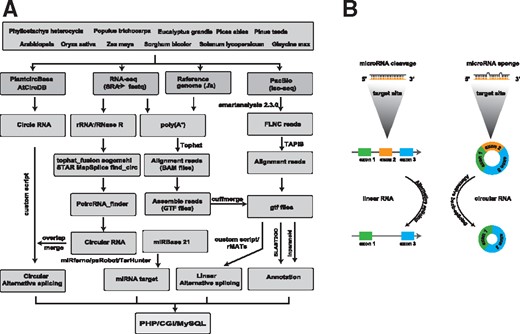
The bioinformatics workflow used for identification of miRNA target sites in splice variants. (A) The workflow includes pipeline for identification of AS from linear and circRNA, miRNA target sites and overlapped regions in splice isoforms. ASmiR consists of MySQL and online submission system. (B) Model of regulation among AS, miRNA and circRNA
For the analysis of PacBio full-length non-chimeric (FLNC) reads, we followed previous method (Wang et al., 2017). In brief, consensusTools from the smrtanalysis_2.3.0 (Pacific Biosciences of California, Inc.) was adopted to get reads of insert. Then, the pbtranscript.py from the smrtanalysis_2.3.0 was used for the full-length read identification. SMRT long reads were corrected by Illumina reads using LSC 1.alpha with Bowtie 2 version 2.2.1 alignment (Au et al., 2012). Then, PacBio sequences were mapped to corresponding genome using GMAP (version 2015-11-20) (Wu and Watanabe, 2005) with following option: -B 5 -K 8000 -t 40 -f 2 -n 1 (Wu et al., 2016). Above GFF3 format was transferred into GTF format using gffread—one of the cufflinks tool (Trapnell et al., 2012). Then AS events were identified based on above GTF file using rMATS (Shen et al., 2014a, b). In this study, we also used TAPIS, which is a reliable pipeline to identify splicing isoforms using full-length cDNA sequences from PacBio SMRT long reads (Abdel-Ghany et al., 2016), which identified AS using SpliceGrapher module (Rogers et al., 2012). In brief, the PacBio sequences were mapped to corresponding genome using GMAP (2016-6-30) with following option: ‘-S –no-chimeras –cross-species –expand-offsets 1 -B 5 -K 8000 –A -f samse’. Then the AS events were obtained by TAPIS to identify four major AS types: intron retention (IR), alternative acceptor sites (AltA), alternative donor sites (AltD) and exon skipping (ES) with the following option: ‘run_tapis.py -v -s 1 -m 25’.
MiRferno from sPARTA toolkit was used for miRNA target prediction with default miRNA/mRNA targets pairing score (Kakrana et al., 2014). In order to improve the reliability of miRNA-mRNA target prediction, psRobot (Wu et al., 2012) was also adopted with the default option. TarHunter is a tool for identification cross-species conserved miRNA targets (Ma et al., 2018). In this study, all the ASR sequences were extracted using custom python script according to the genome coordinates, which were required as -b parameter for TarHunter.pl. All the miRNAs were provided as –q parameter for TarHunter.pl. Then, we performed the TarHunter.pl script with following option: ‘–q species.fa -s miR.txt -b dbs -p 0’ to produce orthologous gene alignment files. These gene alignment files were used to predict orthologous miRNA target sites using TarHunter.pl with this option: ‘–q species.fa -s spe1.txt -b dbs -a ortho_aln_MUSCLE.afa -o output -f 9 -M 9 -T 5 parameters’. To evaluate whether miRNA target sites located in the ASR, customized scripts were developed to identify the overlap between genome coordinate of miRNA target sites and ASR from linear and circular RNA. High-quality vector graphics were drawn using PRAPI to visualize and highlight the interplay (Gao et al., 2017).
2.3 Identification of circular RNAs
The circular exonic RNAs were downloaded from AtCircDB (Ye et al., 2017a, b) and PlantcircBase (Chu et al., 2017). Lariat RNA also plays an important role in plant development (Cheng et al., 2017). Thus, we included circular intronic RNAs also in this study. Circular RNA sequences were mapped to the genome using GMAP with the following option: -B 5 -K 8000 -t 40 -f 2 -n 1 (Wu et al., 2016). Then we obtained the genomic coordinates information around the back-spliced junction for these circular RNAs. In order to filter possible false positives, we only used circular RNAs with the most commonly used splicing signals (GT-AG) since non-canonical splicing sites only comprised a small proportion. Then, the circular RNAs derived from annotated gene structures were preserved for downstream analyses.
In this study, we also used PcircRNA_finder, a circular RNAs prediction algorithm that was specially designed for plants (Chen et al., 2016). PcircRNA_finder required rRNA−/RNase R+ RNA-Seq using paired-end sequencing for identification of circular RNAs in plants (Chen et al., 2016). Thus, we collected all available rRNA−/RNase R+ RNA-Seq for species mentioned in this study, which included Arabidopsis thaliana (Liu et al., 2017; Lu et al., 2017; Ye et al., 2015), Oryza sativa (Lu et al., 2015; Ye et al., 2017a, b), Solanum lycopersicum (Yin et al., 2018), Zea mays (Chen et al., 2018) and Glycine max (Rodrigues et al., 2015). The raw sequence data from SRA archives was first converted to FASTQ using fastq-dump (2.5.7) in SRA Toolkit (https://github.com/ncbi/sratoolkit) and then low-quality reads were filtered out using ht2-filter (1.92.1) from high-throughput quality control (HTQC) package (Yang et al., 2013). The first step for circular RNAs prediction was the identification of back-splicing site. Thus above clean reads were mapped to corresponding genome to identify all back-splice sites with several fusion detection methods, such as Tophat-fusion (Kim and Salzberg, 2011), STAR-fusion (Dobin et al., 2013), find_circ (Memczak et al., 2013), Mapsplice (Wang et al., 2010) and segemehl (Hoffmann et al., 2014). These fusion detection methods were used simultaneously to obtain comprehensive back-splice sites. In brief, STAR (2.5.3a) was adopted to get splicing junction file using parameters ‘–runThreadN 8 –chimSegmentMin 20 –chimScoreMin 1 –alignIntronMax 100000 –outFilterMismatchNmax 4 –alignTranscriptsPerReadNmax 100000 –outFilterMultimapNmax 2 –outFileNamePrefix sti –outSAMtype BAM SortedByCoordinate’. Tophat (2.0.11) and Tophat-Fusion were used for obtaining BAM alignment files by using ‘-a 6 –microexon-search -m 2 -p 50’ and ‘-p 50 –fusion-search –keep-fasta-order –bowtie1 –no-coverage-search’, respectively. BED files were identified from find_circ using unmapped BAM file from bowtie2 with this option ‘–very-sensitive –mm -M20 –score-min=C,-15, 0 -x genome_index -q -U fastq_file’. Mapsplice (2.1.8) was used to identify fusion reads using ‘–non-canonical –fusion-non-canonical –min-fusion-distance 200’. Segemehl (0.2.0-418) with ‘-t 45 -s -S -T -D 2’ option was used for BED files with splice sites information. Then circular RNAs were identified by using above cleaned paired-end reads and PcircRNA_finder (Chen et al., 2016). The back-splicing sites with canonical donor/acceptor sites (GT-AG, GC-AG and AT-AC) within annotated genes were preserved. Candidate circRNAs were identified with the following option: ‘PcircRNA_finder/ecircRNA_finder.pl ./splitmap.txt 20000 Pcirc_finder.gff3 5 5 $genome_fasta $read_length 20 $R1 $R2 1’.
2.4 Database design
ASmiR consists of perl CGI and PHP5 interface for accessing and extracting information from MySQL databases (Fig. 1A), which present the interplay among circRNAs, AS and miRNAs (Fig. 1B). The JBrowse (Skinner et al., 2009) was integrated into ASmiR to visualize different tracks, such as genome reference, GFF, AS and miRNA target sites. Also, users can display custom tracks conveniently by uploading their own files.
3 Results
3.1 Identification of AS events in gymnosperm and angiosperm species
In this study, we collected publicly available RNA-Seq and single-molecule long-read sequencing reads to perform a comprehensive analysis of AS events in eleven plant species. In total, we obtained 178 721, 758 569, 884 638, 146 225 error-corrected FLNC reads from Arabidopsis thaliana (Li et al., 2016a, b, c), Zea mays (Wang et al., 2016), Sorghum bicolor (Abdel-Ghany et al., 2016) and Phyllostachys edulis (Wang et al., 2017), respectively. For PacBio full-length cDNA sequence reads, we also used a module from TAPIS (Abdel-Ghany et al., 2016) to perform AS analysis of Iso-Seq data. In moso bamboo, we obtained a total of 6867 AS events, the number of IR, AltA, AltD and ES were 2759, 1760, 1328 and 1020, respectively. In maize, we detected a total of 53 843 AS events and the number of IR, AltA, AltD and ES were 19 911, 11 261, 9593 and 13 078, respectively. In sorghum, we found a total of 6136 AS events. Overall, IR was the predominant pattern with a total number of 3471 events. AltA ranked as the second number with 1079 events. The number of AltD and ES were 737 and 849, respectively. In Arabidopsis thaliana, we generated a total of 7095 AS events, the number of IR, AltA, AltD and ES were 2706, 2155, 1140 and 1094, respectively. At the same time, 182 RNA-Seq libraries were included for the AS identification using rMATS (Shen et al., 2014a, b). In total, comprehensive transcriptome-wide analysis finally yielded 31 603, 84 875, 42 493, 71 131, 57 544, 19 064, 19 448, 26 894, 17 421, 9074 and 19 854 AS events corresponding to 10 280, 14 689, 10 751, 14 785, 17 568, 7185, 7818, 8782, 7832, 3005 and 3827 genes in Arabidopsis thaliana, Sorghum bicolor, Oryza sativa, Zea mays, Glycine max, Solanum lycopersicum, P.trichocarpa, Eucalyptus grandis, Phyllostachys edulis, Picea abies and Pinus taeda, respectively. All above AS events from TAPIS and rMATs were incorporated into the database that we developed. ASmiR database provides comprehensive AS information for future comparative studies between gymnosperm and angiosperm species.
3.2 Identification of miRNA target sites located in alternatively spliced regions (ASRs) from linear RNAs
One interesting question is how miRNA target sites can be included or excluded by AS among different isoforms. However, the interplay between AS and miRNA has never been systematically investigated. To address this, all miRNA target sites were identified, then genome coordinates corresponding to these miRNA target sites were compared with that of AS regions. In total, 4776, 11 709, 9863, 7997, 18 404, 1573, 3084, 439, 6379, 115, 442 potential target sites were mapped to/aligned with the ASRs in Arabidopsis thaliana, Sorghum bicolor, Oryza sativa, Zea mays, Glycine max, Solanum lycopersicum, P.trichocarpa, Eucalyptus grandis, Phyllostachys edulis, Picea abies and Pinus taeda, respectively. For example, Figure 2 shows 631 miRNA target sites located in 891 ASRs using a score cutoff less than 4.5 in Populus. It is notable that the longer isoforms preferentially harbor the miRNA target site, while the shorter isoforms lack the target site and escape miRNA-mediated regulation. Potri.013G086800 was used as an example to demonstrate the characterization of miRNA target site in ASRs (Supplementary Fig. S1). The long isoform TCONS_00091536 has ptc-miR156g binding site, whereas the short isoforms TCONS_00091533, TCONS_00091534 and TCONS_00091535 produced by AltD lack ptc-miR156g binding site. All the results could easily be searched in ASmiR and viewed in each browser track.
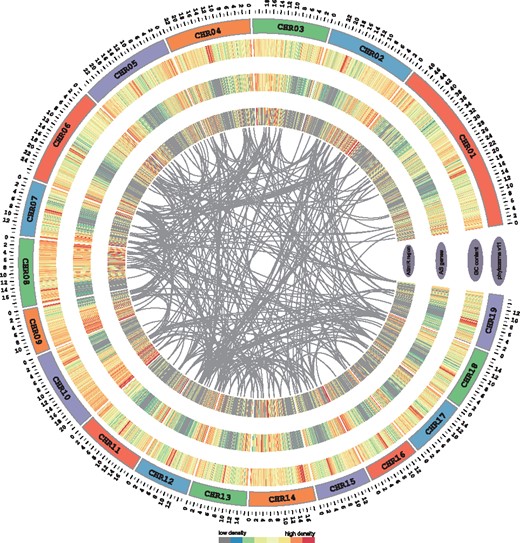
Concentric circles diagram illustrating AS regions with miRNA target sites in P.trichocarpa. From outer to inner circles, it includes a track of phytozome V11 chromosomes and heat map view of total and AS genes density in 100-kb windows, respectively. The inner circle shows the density of ASRs including miRNA target sites. The links between pri-miRNAs and their targets in ASRs are shown as grey lines
In this study, we also added TarHunter's ortho_mode analysis model to predict miRNA target sites in AS regions of eleven species. In total, we identified 407 conserved target sites. These ortholog target sites have been added to the database. Users could determine the corresponding conserved target sites for the retrieved miRNA target sites based on TarHunter prediction.
3.3 Identification of miRNA target sites located in ASRs regions from circular RNAs
In this study, we used circular RNAs with canonical splicing signals (GT-AG) to exclude possible false positives (see methods) from AtCircDB (Ye et al., 2017a, b) and PlantcircBase (Chu et al., 2017). In those two datasets, there are 52 255, 22 556, 874, 2292 and 4810 reliable circular RNA for Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, Zea mays and Glycine max, respectively. The number variation might be caused by different tissue, stage, depth of sequencing and library type, such as RNase R or rRNA-depleted method. In this study, we also identified circRNAs using plant circRNA prediction tool to get reliable and comprehensive candidates (Chen et al., 2016). In total, we obtained 1 850 862 099 paired-end reads. A total of 95 rRNA−/RNase R+ RNA-Seq libraries were obtained from leaves, roots, flowers, seedlings of Arabidopsis thaliana (Liu et al., 2017; Lu et al., 2017; Ye et al., 2015), roots, leaves, ears of Oryza sativa (Lu et al., 2015; Ye et al., 2017a,b), fruits of Solanum lycopersicum (Yin et al., 2018), leaves, ovules, embryo sacs of Zea mays (Chen et al., 2018) and embryos of Glycine max (Ye et al., 2014). We identified 33 538, 7393, 2308, 9587 and 617 circular exonic RNAs by using PcircRNA finder from Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, Zea mays and Glycine max, respectively. Of these, 15 974, 2616, 320, 2139, 27 overlapped with the circular RNA in PlantcirBase (Chu et al., 2017) and ATcircDB (Ye et al., 2017a, b). These circular RNAs represent high reliable ones, which were predicted by multiple circular RNA prediction algorithms. PlantcirBase deposited circular RNAs from stems, roots and mature leaves in soybean (Zhao et al., 2017). However, the raw RNA-Seq dataset from that study was not deposited in SRA. Thus, we used another study in soybean (Rodrigues et al., 2015) for circular RNAs identification using PcircRNA_finder. We only detected 27 overlapped circular RNAs between two studies. The low percentage of overlapping for soybeans between PlantcirBase and PcircRNA_finder suggested the tissues specificity of circular RNAs biogenesis. In order to generate a comprehensive dataset, we merged above circular RNAs identified by PcircRNA finder with AtCircDB (Ye et al., 2017a, b) and PlantcircBase (Chu et al., 2017). After removing the redundant CircRNAs, we obtained 69 631, 27 137, 2855, 9611 and 5340 circular RNAs from Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, Zea mays and Glycine max, respectively. In order to obtain alternative back-splicing events, we used custom scripts for further alternative back-splicing analysis. In total, we identified 62 113, 6069, 418, 2738 and 292 alternative 5’/3’ back-splicing events in Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, Zea mays and Glycine max, respectively. Among these alternative 5’/3’ back-splicing events, 32 811, 6235, 75, 1468 and 575 potential miRNA target sites in ASRs from circular RNAs were identified in Arabidopsis thaliana, Oryza sativa, Solanum lycopersicum, Zea mays and Glycine max, respectively. For example, there were four circular RNAs derived from an RNA-binding protein gene (LOC_Os02g11750) by alternative 5’ back-splicing event (Supplementary Fig. S2). The long circular RNA (chr02: 6063368-6066130) has a target site for miR167d, however, other short isoforms did not have the miRNA target sites.
Subsequently, we constructed a webpage that was dedicated to display information about these circular RNAs and miRNA target information. Through this page, users could get information about genomic location, host gene, organism, circularized exons and algorithms for circular RNAs identification to help researchers to choose reliable candidates circular RNAs.
3.4 Identification of circular RNA overlapping regions within the ASRs from linear RNAs
In Arabidopsis, circRNA from SEPALLATA 3 (SEP3) regulated exon skipping of its own parent genes by forming circRNA-DNA hybrid (Conn et al., 2017). To determine circRNAs located in ASR, customized scripts were developed to identify the overlap between circRNA and ASR from linear RNA. In total, there were 21 633, 7442, 4757, 413, 4668 circRNAs that overlapped with 15 908, 11 015, 9351, 577, 8797 ASRs in Arabidopsis, Oryza sativa, Glycine max, Solanum lycopersicum and Zea mays. ARGININE/SERINE-RICH SPLICING FACTOR 41 (RS41) is a splicing factor, which affects pre-mRNA splicing and biogenesis of a subset of miRNAs (Chen et al., 2015). Pre-mRNAs encoding splicing factors undergo extensive AS (Reddy and Shad Ali, 2011; Zhang et al., 2017a, b). From the assembly data, we found the exon skipping happened in exon 3 (Supplementary Fig. S3). In total, RS41 generated five circRNAs and two of these overlapped with the skipped exon (exon 3). It will be interesting to investigate if these circRNAs modulate the exon skipping of RS41 as reported in the case of SEP3 pre-mRNA splicing. These results provide genome-wide identification of overlapping regions between circRNAs and the ASRs of cognate genes to experimentally investigate the correlation between circular RNAs and AS.
3.5 Implementation of ASmiR as a relational database
ASmiR was developed and implemented in the Linux system with Apache web server. It consists of relational database and web interface designed by PHP to retrieve data from backend MySQL server. For building the ASmiR database, three types of datasets were generated, including linear and circular AS, miRNA target sites and ASRs with miRNA target sites. Importantly, all the datasets can be queried from the easy-to-use web interfaces and are readily accessible to every user. In the current version, users can access the database by several keyword search methods, such as the name of all the types of AS events, gene, genome coordinates, gene orthology, functional categories, etc. Additionally, different threshold criteria can be set to search for miRNA target sites. As shown in Supplementary Figure S4, miR171 was used as the keyword to search for its target genes including ASRs in Phyllostachys edulis (Supplementary Fig. S4A). This query returned one summarized table (Supplementary Fig. S4B), which listed the target genes, coordinates and function description, etc. (Supplementary Fig. S4C). The query results were hyperlinked to GO term, functional description, graphical visualization, sequences of the splice variants and other features (Supplementary Fig. S4D). The user could easily find which exon could generate circular RNA using back-splicing. In addition, users could also visually observe which exon from circular RNAs could be targeted by miRNA. Users could also search gene name directly, and it would return whether there was any back-splicing on the gene. Thus, it provided useful information for experimental design to perform exon-swap experiments.
3.6 Web server for online submission
Because of the availability of sequence data from only a few plants and few tissues and developmental stages, many miRNA-mediated regulatory mechanisms that are development-, tissue- and/or species-specific are yet to be discovered. The web-based tools that we developed can be used to address this as researchers obtain more sequencing data from other plants, tissues and developmental stages. Aside from data deposited into the ASmiR database, users can submit their own small RNA datasets to the online web service, which was constructed based on the CGI. Users can select different cutoff values and paste their own FASTA sequences at the online submission interface shown in Supplementary Figure S5A. Once ASmiR receives the request, it will calculate the miRNA target sites located in the AS region and generate a summary page (Supplementary Fig. S5B) with hyperlinks to a more detailed table and graphical results (Supplementary Fig. S5C and D). Users can also provide both miRNA sequences and splice isoform sequences to search for the potential interplay between miRNA and AS, which is useful for the analysis of sequence data from any species. In summary, this online web service serves as a powerful and easy-to-use web resource for the community without the computational infrastructure to analyze the interplay between miRNAs and AS with users’ own high throughput sequencing data.
4 Conclusion
At present, the interplay between AS and miRNA target sites remains to be elucidated in plants, thus it is important to determine which splice variants are targeted by miRNA and to gain a comprehensive global view of this regulation, and its contribution to the post-transcriptional regulation of gene expression. The alternatively spliced events of the linear and circRNAs were collected using the up-to-date SMRT isoform sequencing and Illumina sequencing data. ASmiR serves as a central resource for integrating AS events from linear and circular RNAs, miRNA targets and the interplay between two important post-transcriptional regulatory processes of gene expression. More importantly, this online-tool with the user-friendly interface allows to identify miRNA target sites located in the alternatively spliced regions of linear and circRNAs using user-defined datasets for any species.
ASmiR also provides many additional tools including BLAST of AS region, extracting sequences and feature description using Perl CGI-script. Besides, ASmiR also documents the miRNA precursor including AS events and splicing factor genes targeted by miRNAs. In summary, ASmiR is a powerful platform for studying the interplay between miRNA and AS for model plants, woody perennials and any plant species. This database and online-tools minimize the bioinformatics skills needed for the end-users and allows them to focus on biological functions.
These AS events are still far from saturation because more AS events will be detected with more spatiotemporal transcriptomes. Since ASmiR is an ongoing curation, it will be continuously maintained and updated as the new data are published. More PacBio-generated full-length isoform sequencing, RNA-Seq and circular RNA sequence resource will continue to be collected and integrated into ASmiR. It is anticipated that this resource will provide further insights into the post-transcriptional regulation of gene expression, which will accelerate domestication and genome-assisted breeding of crop plants and woody perennials in future.
Acknowledgements
We give special thanks to Blake C. Meyers and Atul Kakrana at Donald Danforth Plant Science Center for help on miRferno script.
Funding
This work was supported by the National Natural Science Foundation of China Grant (Grant No. 31570674 and 31800566), the National Key R&D Program of China (2018YFD0600101 and 2016YFD0600106), the International Science and Technology Cooperation and Exchange Fund from Fujian Agriculture and Forestry University (KXGH17016), Natural Science Foundation of Fujian Province of China (Grant No. 2018J01608) and Program for scientific and technological innovation team in university of Fujian province (No. 118/KLA18069A).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Huiyuan Wang, Huihui Wang and Hangxiao Zhang authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}