





Abstract
In this paper, we present an open source package with the latest release of Evolutionary-based BIClustering (EBIC), a next-generation biclustering algorithm for mining genetic data. The major contribution of this paper is adding a full support for multiple graphics processing units (GPUs) support, which makes it possible to run efficiently large genomic data mining analyses. Multiple enhancements to the first release of the algorithm include integration with R and Bioconductor, and an option to exclude missing values from the analysis.
Evolutionary-based BIClustering was applied to datasets of different sizes, including a large DNA methylation dataset with 436 444 rows. For the largest dataset we observed over 6.6-fold speedup in computation time on a cluster of eight GPUs compared to running the method on a single GPU. This proves high scalability of the method.
The latest version of EBIC could be downloaded from http://github.com/EpistasisLab/ebic. Installation and usage instructions are also available online.
Supplementary data are available at Bioinformatics online.
1 Introduction
Biclustering is an unsupervised machine learning technique which attempts to detect meaningful data patterns that are distributed across different columns and rows of the input dataset. This allows biclustering to capture heterogeneous patterns that manifest only in subsets of genes and subsets of samples. Biclustering has been commonly applied to genomic datasets (Padilha and Campello, 2017) and has proven to be successful in revealing potential diagnostic biomarkers (Liu et al., 2018), and claimed to identify altered transcriptional profiles in breast cancer (Singh et al., 2018).
With exponentially increasing sizes of the input datasets, there is an emerging need for effective and efficient methods that would scale well with growing amounts of data. Although there was discussion on possibility of applying biclustering to larger datasets (Kasim et al., 2016; Padilha and Campello, 2017), hardly any biclustering study involved large genomic dataset. This motivated the emergence of parallel biclustering method. Some of the most recent parallel methods use multiple threads—e.g. runibic (Orzechowski et al., 2017), or Message Passing Interface—e.g. ParBiBit (González-Domínguez and Expósito, 2018), or graphics processing unit (GPU)—e.g. CCS (Bhattacharya and Cui, 2017).
One of the recent advancements in biclustering area was introduction of Evolutionary-based BIClustering (EBIC), which takes advantage of multiple other evolutionary computation strategies (Orzechowski et al., 2018a, b). This representative of hybrid biclustering algorithms (Orzechowski and Boryczko, 2016a, b, c) has been shown to outperform multiple state-of-the-art methods in terms of accuracy. Although the original concept of EBIC provided theoretical support for multiple GPUs, all the previous evaluations have been made using a single GPU. Thus, the rationale of involving multiple GPUs was not clear. Another constraint for EBIC was hardware limitation of the size of the dataset to 65 535 rows per GPU. This required large clusters of GPUs in order to run analyses and greatly restricted application of the method.
In this paper, we introduce the open source package built on top of the upgraded version of the method. First and foremost, a full support for multi-GPUs is added, which allows to analyze datasets with almost unlimited numbers of rows (available memory of devices are the only constraint). Second, an integration with Bioconductor was added, which enables the user to run all the analysis from the R level. Third, a different method for performing analysis was added, which depends on the presence or absence of missing values within the data. Last, but not least, some bugs have been fixed and optimizations were made for more efficient memory management. All above combined make this open source software ready out-of-the-box or big data biclustering analyses.
2 Materials and methods
The major objective of EBIC is to make data analyses as easy as possible. Thus, in its simplest form, the method requires only a single parameter, which is a dataset in a popular comma-separated values format. Running the algorithm on multiple GPUs requires to specify additional parameters with number of required devices. An example commands to run the software are as follows (the second one runs the method on 4 GPUs):
./ebic -i input.txt
./ebic -i input.txt -g 4
After a proper installation, the method may also be run within R environment, using system function, which points to the location of EBIC binary. For detailed information on command line parameters, and general information on software installation and usage please refer to Supplementary Material as well as online documentation available on Github.
The new version of EBIC introduced herein provides a comprehensive open source framework for performing biclustering analysis. The major improvements over the original release of the method include:
Support for Big Data. In the previous version of EBIC only a very limited number of rows could be processed on a single GPU. Kernel grid constrained the maximal number of rows analyzable by an EBIC to 65 535 per GPU. Thus, at least eight GPUs were needed to analyze large datasets, e.g. modern methylation datasets. Our new implementation overcomes this limitation, allowing to analyze up to 231–1 rows per single GPU (devices with computing capabilities 3.0). This greatly enhances the flexibility and applicability of the method to almost any type of data. This comes at a cost of reducing the size of genetic algorithm population down to 65 535. This remains a large number, as for the majority of genomic datasets the algorithm converged using a population size of 1600 or less individuals.
Handling missing values. We introduce a very important feature which allows to remove the impact of missing values on the results of the method. As EBIC search is driven by counting of rows, a greater or equal relation between the values in columns used to capture missing values, instead of the real trends in the data. This posed a drawback, especially for datasets with high percentage of missing values. Instead of finding useful patterns in the data, EBIC used to become more attracted in detecting the emptiness. In the current release, missing values might be replaced with a pre-defined value (e.g. 0 or 999) passed as an input parameter. This value no longer counted toward the score of the bicluster. Thus, the method is more focused on detecting the trends, instead of emptiness. Please be aware that performing a dataset normalization could impact the representation of missing values in each column if missing values are involved in calculations.
Different input file formats support. EBIC allows different delimiters in input file, which simplifies portability of data between R and EBIC. The input datasets needs to meet the following restrictions: (i) the data values might be separated by either comma, tabulator, space or semicolon, (ii) the labels of rows and columns need to be present (in left-most column and top-most row) and (iii) the left- and top-most field representing crossing of columns and rows labels needs not to be empty.
Compatibility with R and Bioconductor. The results returned by EBIC could be easily saved into a format loadable by Bioconductor R package biclust in order to perform biological validation. In Supplementary Material we provide detailed workflow presenting how to use EBIC, all within R environment. Notice that EBIC is not a part of Bioconductor and it needs to be installed separately.
Workflow for methylation data analysis. EBIC was capable to capture bio-meaningful signals in methylation data. A tutorial is presented in a Supplementary Material.
3 Results
In order to assess running times of the algorithm we have performed tests on from one up to eight GPUs on datasets with varying number of rows and columns. The Gene Expression Omnibus accession numbers of the datasets as well as run times of the algorithm are presented in Table 1.
Datasets used in the experiment as well as an average running time (in minutes) using a cluster of eight GeForce GTX 1080 Ti GPUs
| Dataset | Genes | Samples | Description | Run time (min) |
|---|---|---|---|---|
| GDS1490 | 12 483 | 150 | Neural tissue profiling | 7.1 |
| GSE65194 | 54 675 | 178 | Breast cancer | 18.3 |
| GSE84493 | 436 444 | 310 | Prostate cancer methylation | 24.5 |
| Dataset | Genes | Samples | Description | Run time (min) |
|---|---|---|---|---|
| GDS1490 | 12 483 | 150 | Neural tissue profiling | 7.1 |
| GSE65194 | 54 675 | 178 | Breast cancer | 18.3 |
| GSE84493 | 436 444 | 310 | Prostate cancer methylation | 24.5 |
Datasets used in the experiment as well as an average running time (in minutes) using a cluster of eight GeForce GTX 1080 Ti GPUs
| Dataset | Genes | Samples | Description | Run time (min) |
|---|---|---|---|---|
| GDS1490 | 12 483 | 150 | Neural tissue profiling | 7.1 |
| GSE65194 | 54 675 | 178 | Breast cancer | 18.3 |
| GSE84493 | 436 444 | 310 | Prostate cancer methylation | 24.5 |
| Dataset | Genes | Samples | Description | Run time (min) |
|---|---|---|---|---|
| GDS1490 | 12 483 | 150 | Neural tissue profiling | 7.1 |
| GSE65194 | 54 675 | 178 | Breast cancer | 18.3 |
| GSE84493 | 436 444 | 310 | Prostate cancer methylation | 24.5 |
EBIC obtained up to 6.6× fold speedup using eight GPUs on a dataset with over 436 k rows. For datasets with smaller number of rows, the speedups were around 1.2× (12 k rows) and 2.75× (55 k rows). The relation between the different number of GPUs used and obtained speedup is presented in Figure 1.
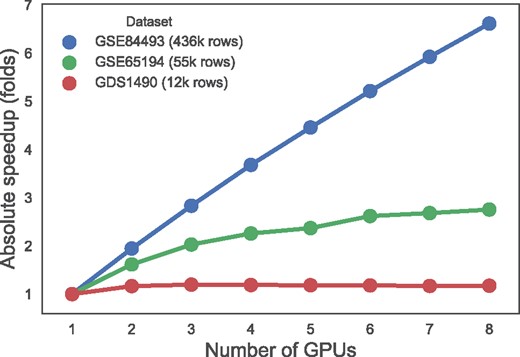
Since its proposal EBIC was applied to numerous datasets: both synthetic and real genomic ones. It was found to be very robust in detecting correlated genomic patterns. EBIC was found to be especially helpful in detecting large biclusters, in which multiple rows are correlated with each other within some subset of columns. We believe that EBIC could serve as a useful tool for performing exploratory analysis of correlations of rows within the data.
4 Conclusions
In this paper, we present the recent advancements in one of the leading biclustering methods. The algorithm was wrapped into a framework, which is conveniently integrated with R and allows multiple input file formats. In Supplementary Material we also demonstrate that even for such a large genomic dataset, the results provided by EBIC are bio-meaningful. We conclude that EBIC, released as open source package, is a very convenient tool for getting insight from large genomic datasets.
Future work on EBIC involves developing a better initialization method of the initial population, to make a method more robust for wide biclusters. We also consider integrating expert knowledge into a software. As running EBIC on some of the datasets may result in highly overlapping biclusters, we also consider modifications to the list storing the best found solutions in order to limit the overlap between the biclusters.
Funding
This research was supported in part by PL-Grid Infrastructure and by grants [LM012601, TR001263, ES013508] from the National Institutes of Health (USA).
Conflict of Interest: none declared.
References


{kind=link}