





Abstract
Mobile element insertion (MEI) is a major category of structure variations (SVs). The rapid development of long read sequencing technologies provides the opportunity to detect MEIs sensitively. However, the signals of MEI implied by noisy long reads are highly complex due to the repetitiveness of mobile elements as well as the high sequencing error rates. Herein, we propose the Realignment-based Mobile Element insertion detection Tool for Long read (rMETL). Benchmarking results of simulated and real datasets demonstrate that rMETL enables to handle the complex signals to discover MEIs sensitively. It is suited to produce high-quality MEI callsets in many genomics studies.
rMETL is available from https://github.com/hitbc/rMETL.
Supplementary data are available at Bioinformatics online.
1 Introduction
Mobile element insertions (MEIs) represent about 25% of structure variations (SVs) in human genomes (Gardner et al., 2017), which are mainly contributed by active transposons such as Alu, L1 and SVA families (Stewart et al., 2011). Efforts have been made to detect MEIs with short reads (Sudmant et al., 2015), however, short-read-based approaches have their own limitations to deal with repetitive mobile elements.
Long reads are promising to handle repeats and more sensitively detect SVs (Sedlazeck et al., 2018a). However, with the repetitiveness of mobile elements and high sequencing error rates, the MEI signals implied by long reads are highly complex. State-of-the-art long-read-based SV detection tools use unified approaches to detect various kinds of SVs (Sedlazeck et al., 2018b). However, this ‘one-fits-all’ strategy does not fully consider the characteristics of MEIs, which may affect the detection.
Herein, we propose Realignment-based Mobile Element insertion detection Tool for Long read (rMETL). rMETL takes advantage of its specifically designed chimeric read re-alignment approach to handle the complex MEI signals. This novel approach has improved ability to produce high quality MEI callsets.
2 Materials and methods
Using aligned long reads (a sorted BAM file) as input, rMETL extracts and re-aligns chimerically aligned reads to discover MEIs (range from 50 bp to 1 million bp) in four steps.
rMETL extracts the chimerically aligned parts of the reads which have split alignment, large clippings and/or large indels;
rMETL clusters the chimerically aligned read parts in pre-defined rules to infer a set of putative MEI sites as candidates;
rMETL realigns the clustered read parts to the consensus sequences of Alu, L1 and SVA families with a well-tuned aligner;
rMETL investigates the realignment results to find out the evidence to call MEIs as well as filter false positive candidates.
Please also refer to Supplementary Figures S1 and S2 for schematic illustrations and Supplementary Notes for more detailed information on the implementation of rMETL.
3 Results
We implemented rMETL on simulated and real datasets to assess its ability. A state-of-the-art long-read-based SV calling approach, Sniffles (Sedlazeck et al., 2018b), was employed for comparison.
Four PacBio-like datasets (mean read length: 8000 bp, mean error rate: 15%) on four sequencing depths (5×, 10×, 20× and 50×) were simulated with an in silico haploid human genome having 20 000 MEIs (Section 2.1 of Supplementary Notes). For both rMETL and Sniffles, all the parameters were set as default values except the numbers of supporting reads (−s parameters), which were tuned as 5 for the 5×, 10× and 20× and 10 for the 50× datasets, referring to previous studies on the tradeoff between sensitivity and specificity (Sedlazeck et al., 2018b).
The sensitivities and accuracies of rMETL and Sniffles are in Table 1 and Supplementary Table S1. Overall, rMETL achieves higher sensitivity than Sniffles, especially on the lower depth (5× and 10×) datasets. Moreover, both of the two approaches have low false positive rates (0.01–0.23% for rMETL and 0.04–1.95% for Sniffles).
Sensitivities of rMETL and Sniffles on four simulated PacBio datasets (−s indicating the number of supporting reads parameter)
| 5× (−s 5) | 10× (−s 5) | 20× (−s 5) | 50× (−s 10) | |
|---|---|---|---|---|
| rMETL | 49.24% | 78.64% | 88.19% | 90.14% |
| Sniffles | 28.06% | 68.93% | 86.19% | 89.43% |
| 5× (−s 5) | 10× (−s 5) | 20× (−s 5) | 50× (−s 10) | |
|---|---|---|---|---|
| rMETL | 49.24% | 78.64% | 88.19% | 90.14% |
| Sniffles | 28.06% | 68.93% | 86.19% | 89.43% |
Sensitivities of rMETL and Sniffles on four simulated PacBio datasets (−s indicating the number of supporting reads parameter)
| 5× (−s 5) | 10× (−s 5) | 20× (−s 5) | 50× (−s 10) | |
|---|---|---|---|---|
| rMETL | 49.24% | 78.64% | 88.19% | 90.14% |
| Sniffles | 28.06% | 68.93% | 86.19% | 89.43% |
| 5× (−s 5) | 10× (−s 5) | 20× (−s 5) | 50× (−s 10) | |
|---|---|---|---|---|
| rMETL | 49.24% | 78.64% | 88.19% | 90.14% |
| Sniffles | 28.06% | 68.93% | 86.19% | 89.43% |
Furthermore, we implemented rMETL on a 50× simulated PacBio-like dataset from another in silico haploid human genome having 20 000 non-MEI insertions (Section S2.1 of Supplementary Notes). Only 366 (1.8%) of the 20 000 events were false positively called as MEIs, suggesting that rMETL has the ability to prevent false positives.
rMETL and Sniffles were further benchmarked with a 55× real PacBio dataset (Zook et al., 2014) and a 28× real ONT dataset (Jain et al., 2018). Their -s parameters were respectively set as 10 (PacBio) and 5 (ONT), referring to the previous study (Sedlazeck et al., 2018b). A callset proposed by 1000 Genomes Project (Sudmant et al., 2015) (which is produced by multiple approaches) and other four callsets generated by state-of-the-art short-read-based MEI calling tools, i.e. MELT (Gardner et al., 2017), Tangram (Wu et al., 2014), Mobster (Thung et al., 2014) and Tea (Lee et al., 2012), were employed as pseudo ground truth. Each of them is termed as a ‘SR-callset’.
rMETL called 4704 and 5439 MEIs, and Sniffles called 21613 and 59870 INS/DELs, on the PacBio and ONT datasets respectively. Sniffles’ higher numbers of calls are also reasonable since it detects all kinds of large insertions and deletions. We assessed the numbers of the calls supported by various SR-callsets and observed two issues.
1) It indicates that, the callsets of rMETL covered 1589 (with the PacBio dataset) and 1588 (with the ONT dataset) of the 1628 MEIs which co-exists in at least two SR-callsets (Fig. 1 and Supplementary Table S2). Moreover, the upset plots (Supplementary Figs S3–S6) indicate that rMETL recovered 1696 (with the PacBio dataset) and 1699 (with the ONT dataset) of 1764 MEIs in the 1000 Genomes Project callset. This suggests that in absolute terms rMETL has good sensitivity, considering that the MEIs called by multiple approaches could be more confidently seen as true MEIs, and rMETL recovered most of them.
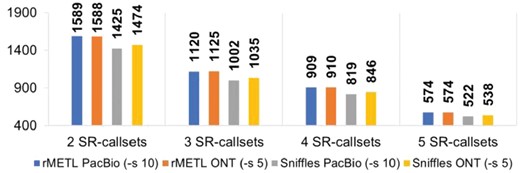
The numbers of long read-based calls supported by various numbers of SR-callsets. Each bar indicates a specific callset produced by rMETL or Sniffles on PacBio or ONT dataset, and its height indicates the number of calls in the callset being supported by X (2–5) SR-callsets, i.e. the calls also exist in at least X SR-callsets
2) On the same levels of SR-callset supports (i.e. supported by the same numbers of SR-callsets), rMETL always has more MEI calls than Sniffles does (Fig. 1). This indicates that the sensitivity of rMETL is higher than that of Sniffles.
We find that the good sensitivity of rMETL derives from its realignment approach, which enables to transform ambiguous and chimeric read alignments into homogenous alignments. This helps to find strong MEI evidence from complex signals, which is still non-trivial to unified SV detection approaches. An example is in Supplementary Figure S7.
There are also MEIs called by rMETL which are not supported by any of the SR-callsets (i.e. 2412 and 3120 calls for the PacBio and ONT datasets, respectively). However, 77% (PacBio) and 79% (ONT) of such calls also exist in the callset of Sniffles (Supplementary Fig. S8), indicating that they could be plausible. We found that most of such unsupported calls also have strong evidence. That is, there are many chimeric read parts in the called MEI regions, and most of them can be confidently aligned to mobile elements (Supplementary Fig. S9).
The elapsed times, CPU times and memory footprints with 1, 2, 4, 8 and 16 CPU threads were assessed (Supplementary Table S3). Mainly, rMETL processed the PacBio and the ONT datasets in respectively 2.1 and 1.5 h with 8 CPU threads (peak memory: 7.05 and 6.52 GB), about 2 times faster than Sniffles.
The benchmarking results suggest that overall rMETL has good ability to detect MEIs. However, it has a few drawbacks. rMETL might fail at the incorrect realignment of read parts or the lack of supporting reads. These are also important future works for us to improve rMETL. Moreover, to some extent, rMETL relies on the consensus sequences of mobile elements. A more detailed discussion is in Supplementary Notes.
Funding
This work was partially supported by the National Key Research and Development Program of China (Nos: 2018YFC0910504, 2017YFC0907503 and 2017YFC1201201).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Tao Jiang and Bo Liu authors should be regarded as Joint First Authors.


{kind=link}