





Abstract
Searching for local sequence patterns is one of the basic tasks in bioinformatics. Sequence patterns might have structural, functional or some other relevance, and numerous methods have been developed to detect and analyze them. These methods often depend on the wealth of information already collected. The explosion in the number of newly available sequences calls for novel methods to explore local sequence similarity. We have developed a new method for iterative motif scanning that will look for ungapped sequence patterns similar to a submitted query. Using careful parameter estimation and an adaptation of a fast string-matching algorithm, the method performs significantly better in this context than the existing software.
The IGLOSS web server is available at http://compbioserv.math.hr/igloss/.
Supplementary data are available at Bioinformatics online.
1 Introduction
Motif scanning methods are at the heart of many bioinformatics procedures. For example, secondary structure recognition, proteome annotation and, in general, protein family assignment (Bateman et al., 2004), all depend—to a certain extent—on detecting a variant of a (amino acid) motif in a given sequence or a set of sequences. Consequently, numerous methods—Smith-Waterman algorithm (Waterman et al., 1976), BLAST (Altschul et al., 1990), PSSM (Gribskov et al., 1987), Viterbi (Viterbi, 1967)—and applications—e.g. FIMO (Grant et al., 2011)—have been developed. Typically, the application takes a motif profile as an input, and then, using a dynamic programming approach, or some approximation, finds a version of optimal local alignment in each scanned sequence. Clearly, accuracy of this approach depends—among other things—on the motif profile being of a sufficient quality and size. A considerable increase in the number of newly available sequences, where only a small portion has been properly analyzed, makes the task of creating an unbiased, representative profile increasingly difficult, and necessitates a different approach.
In this note, we present IGLOSS—iterative gapless local similarity search—a web-application that will, in a proteome or a collection of proteomes, find sequence patterns similar to a submitted query. The query can consist of one or more sequences of equal length, the level of required similarity can be easily controlled, and we provide simple options for conserved/neutral positions, as well.
2 Implementation
Our iterative approach is implemented in a straightforward fashion: initially, a crude profile—with crudeness depending on the size of the query—is built, and the dataset is scanned, with the maximal log-odds score reported for each sequence. Standard mathematical results (see Supplementary Material) guarantee that the scores will be approximately logistically distributed, and motifs with scores above the predetermined scale are used to build a new profile. Clearly, this procedure stops when there is no change in the list of positives, or the predetermined number of iterations is reached.
3 Example and evaluation
We applied our server to the GDSL-lipase protein family from five higher plants—Arabidopsis thaliana (AT) (TAIR9), Oryza sativa (OS) (MSU v7), Solanum tuberosum (ST) (ITAG1), Solanum lycopersicum (SL) (ITAG2.3) and Beta vulgaris (BV) (KWS2320). Proteins in this family display fairly low overall sequence similarity, but are reasonably well described by the presence of five characteristic motifs, also called blocks (Vujaklija et al., 2016). The evaluation consisted of submitting a single sequence, typical for block I, to our scanner and checking the annotation of sequences in which positive hits are found. Annotation was determined by processing the information from GoMapMan resource (Ramšak et al., 2014). We measure the quality of our results by computing true positive rate (TPR), i.e. sensitivity, and positive predictive value (PPV), i.e. precision, and compare them to those obtained by iterative BLAST (PB) (i.e. PSI-BLAST, Altschul et al., 1997) and iterative HMMer (JH) (Finn et al., 2015), using the same input. We ran IGLOSS on each organism at 35 different levels of the scale parameter, and PB and JH for 25 and 35 levels of e-value, respectively, to obtain matching PPVs. Cumulative results of these tests are presented with PPV-TPR curves below (note that for statistical reasons—explained in the Supplementary Material—the usual ROC curve is not suitable in this situation):
As can be seen from Figure 1, IGLOSS—in terms of accuracy—outperforms PB, more-or-less matches JH for values of PPV below 0.7, and has a considerably higher sensitivity when precision is above 0.7. While PSI-BLAST and jackHMMer are both more versatile and much faster applications, our decrease in speed is accompanied by a significant increase in accuracy. It should also be pointed out that a comparison with non-iterative methods would be unfair. The mathematical background of our method is very similar to that of BLAST and HMMer, with main differences in implementation being purpose-specific—that is, iterative-specific—model building and the distribution parameter estimation. While this is certainly time consuming—especially the latter—it appears that it contributes towards considerably greater accuracy.
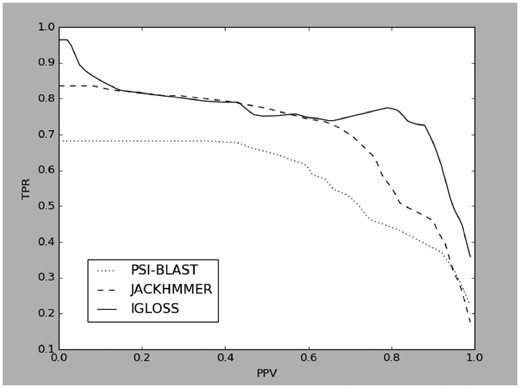
PPV-TPR curve for IGLOSS, PSI-BLAST and jackHMMer
In conclusion, we suggest that our method is a viable alternative when it comes to motif scanning, protein family analysis, and even proteome annotation. On the other hand, while IGLOSS can be used as a fast iterative motif scanner, its primary aim is to help researchers explore common sequence patterns in a proteome.
Funding
This work was partially supported by the European Regional Development Fund [KK.01.1.1.01.0009—DATACROSS] (SR), the Slovenian Research Agency [J7-7303] (MZ) and the Croatian Science Foundation [IP-2014-09-2285] (BR), [IP-2016-06-1046] (PG) and [IP-2018-01-7317] (SR).
Conflict of Interest: none declared.
References


{kind=link}