





Abstract
Hidden Markov models (HMMs) and profile HMMs form an integral part of biological sequence analysis, supporting an ever-growing list of applications. The aphid R package can be used to derive, train, plot, import and export HMMs and profile HMMs in the R environment. Computationally-intensive dynamic programing recursions, such as the Viterbi, forward and backward algorithms are implemented in C++ and parallelized for increased speed and efficiency.
The aphid package is released under the GPL-3 license, and is freely available for download from CRAN and GitHub (https://github.com/shaunpwilkinson/aphid).
Supplementary data are available at Bioinformatics online.
1 Introduction
Hidden Markov models (HMMs) underlie many of the most important tasks in computational biology, including sequence alignment, trimming and annotation, gene discovery and database searching. Originally developed for speech recognition, their application has had profound impacts in molecular biology, facilitating full probabilistic analysis in place of heuristic approximation. Pioneering this transition are two groups lead by Anders Krogh and Sean Eddy, whose respective software packages SAM (https://compbio.soe.ucsc.edu/sam.html) and HMMER (Eddy, 2009) have underpinned HMM-based bioinformatic analysis for over two decades.
Bioinformatic tasks are increasingly carried out in the R environment (R Core Team, 2018), aided by the reproducible code-based workflow and cross-platform availability of a wide range of open-source packages with an active support community. However, to date there are only a handful of packages dedicated to HMMs, none of which support profile HMMs, a variant commonly used in biological sequence analysis. Here, we present the aphid R package for analysis with profile HMMs. The package contains functions for deriving, training, plotting, importing and exporting both standard and profile HMMs, as well as implementations of the forward, backward, Viterbi and maximum a posteriori algorithms. The recursive computations are implemented in C++ via the Rcpp package (Eddelbuettel and Francois 2011), and parallelized for increased speed and efficiency.
2 Implementation
A standard discrete HMM is a probabilistic data-generating mechanism for a sequence or set of sequences. It is depicted by a network of states each emitting symbols from a finite alphabet according to a set of emission probabilities, whose values are specific to each state. The states are traversed by an interconnecting set of transition probabilities, that includes the probability of remaining in any given state and those of transitioning to each of the other connected states. Profile HMMs expand on HMMs to provide a position-specific scoring system that can account for substitutions, gaps and insertions, making them well suited to biological sequence analysis (Krogh et al., 1994). The precursor to a profile HMM is usually an MSA. Each column in the alignment will often (but not always) be represented by one internal position or module in the model, with each module consisting of three states: a silent delete state that does not emit residues, an insert state with emission probabilities reflecting the background residue frequencies of the entire alignment and a match state with emission probabilities reflecting the residue frequencies in the specified alignment column. These states are depicted in Figure 1 as circles, diamonds and rectangles, respectively.
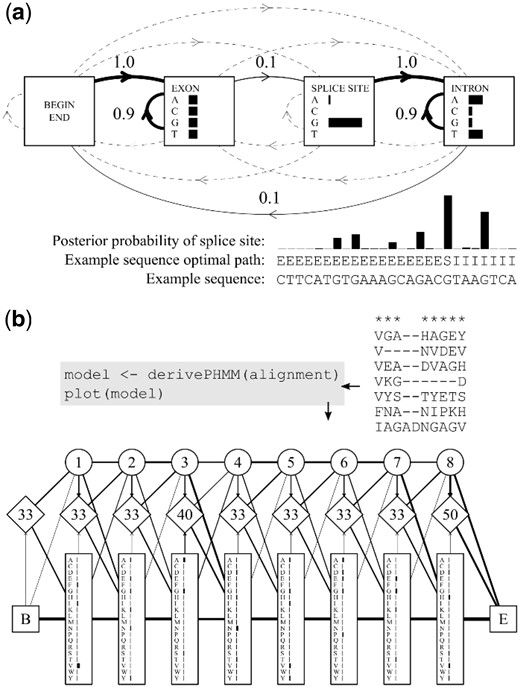
Examples of standard (a) and profile (b) HMMs derived and plotted with aphid. Horizontal bars show emission probabilities and line weights represent transition probabilities (dashed lines show probabilities of 0). Module numbers are shown in the profile HMM delete states (circles) and insert–insert transition probabilities in the insert states (diamonds). Examples modified from Eddy (2004) and Durbin et al. (1998)
The aphid package introduces two primary object classes, HMM and PHMM for standard and profile HMMs, generated using the functions deriveHMM and derivePHMM, respectively. These objects are lists containing emission and transition probability matrices (elements named E and A), vectors of background emission and transition probabilities (qe and qa, respectively) and other non-mandatory model metadata.
To derive a profile HMM, either an MSA or a list of sequences is passed to derivePHMM. Multiple strategies are supported for marking alignment columns as arising from match or insert states, including gap frequency thresholding, alignment inheritance and the maximum a posteriori method outlined in Durbin et al. (1998).
The train function optimizes model parameters using the Baum Welch or Viterbi training algorithm. This method is employed by the align function to produce high-quality MSAs via iterative model training and re-alignment. If no model is provided, align first derives a preliminary PHMM from a seed sequence (selected based on minimum sequence weight to avoid outliers, and treated as a single-row alignment) and refines the model on the full sequence set (using either Baum Welch or Viterbi training, specified with the method argument). The sequences are then aligned to the model to produce an MSA. If only two sequences are present in the input list, the function performs a pairwise alignment without a profile HMM (i.e. a Smith–Waterman or Needleman–Wunch alignment). Model training and alignment operations can be run on multiple processors by setting the cores argument to 2 or more.
To maintain compatibility between aphid and other software, profile HMMs can be exported as text files in the HMMER v3 format (http://www.hmmer.org/) using the function writePHMM. Similarly, a HMMER v3 text file can be parsed into R as an object of class PHMM with the readPHMM function. Utility functions are also provided for plotting HMMs and profile HMMs, calculating posterior probabilities, sequence simulation, sequence weighting and several other tasks.
aphid is designed to be used in conjunction with the ape package (Paradis et al., 2004), whose binary DNAbin and AAbin object types are recommended over standard character sequences for speed and efficiency. However, the aphid package also supports standard characters, making it compatible with applications outside of biological sequence analysis.
3 Examples
Eddy (2004) provides an example of a toy HMM for finding single-nucleotide splice sites located between gene exons and introns. Given that introns and exons have different guanine-cytosine content, and the intervening splice site is generally a guanine, we can calculate the optimal sequence of hidden states (and the associated likelihood) for any given sequence using the Viterbi algorithm (Fig. 1a). We can also use the forward and backward algorithms for posterior decoding, to find the probability for each residue as a splice site candidate (Fig. 1a). This example demonstrates the flexibility and utility of standard HMMs for gene-annotation applications. Durbin et al. (1998) provide a small globin sequence alignment to derive a simple amino acid profile HMM (Fig. 1b). In this case, 8 of the 10 alignment columns are marked as sufficiently conserved to be assigned modules in the model according to the maximum a posteriori algorithm [the aphid default method; residues in columns 4 and 5 are considered to have been emitted from the insert state of module 3; Durbin et al. (1998)]. The residue emission frequencies depicted within each module reflect those of the respective alignment columns, with background pseudo-counts added to avoid zero-value emission probabilities. Transition probabilities are also calculated from the marked alignment columns, and are corrected in the same manner (depicted by weighted lines in the graph; Fig. 1b). Source code for these examples and benchmarking results are available in the Supplementary Script provided. Additional details and explanations can be found in the package vignette (https://cran.r-project.org/package=aphid).
4 Conclusion
Programs such as SAM and HMMER have facilitated probabilistic sequence analysis and improved inference for bioinformatic applications. The aphid package builds on these resources, providing the flexibility to work with profile HMMs in the R environment. The package is under active development, and feedback from the community is appreciated.
Funding
S.P.W. was supported by a Rutherford Foundation Postdoctoral Fellowship from the Royal Society of New Zealand (grant number RFT-VUW1501-PD).
Conflict of Interest: none declared.
References
R Core Team (


{kind=link}