





Abstract
Despite intense effort, it has been difficult to explain chaperone dependencies of proteins from sequence or structural properties.
We constructed a database collecting all publicly available data of experimental chaperone interaction and dependency data for the Escherichia coli proteome, and enriched it with an extensive set of protein-specific as well as cell-context-dependent proteostatic parameters. Employing this new resource, we performed a comprehensive meta-analysis of the key determinants of chaperone interaction. Our study confirms that GroEL client proteins are biased toward insoluble proteins of low abundance, but for client proteins of the Trigger Factor/DnaK axis, we instead find that cellular parameters such as high protein abundance, translational efficiency and mRNA turnover are key determinants. We experimentally confirmed the finding that chaperone dependence is a function of translation rate and not protein-intrinsic parameters by tuning chaperone dependence of Green Fluorescent Protein (GFP) in E.coli by synonymous mutations only. The juxtaposition of both protein-intrinsic and cell-contextual chaperone triage mechanisms explains how the E.coli proteome achieves combining reliable production of abundant and conserved proteins, while also enabling the evolution of diverging metabolic functions.
The database will be made available via http://phdb.switchlab.org.
Supplementary data are available at Bioinformatics online.
1 Introduction
Protein folding is thought to be thermodynamically determined, and thus to be a spontaneous reaction toward the most stable conformational ensemble (Hartl and Hayer-Hartl, 2009; Itzhaki and Wolynes, 2008). However, protein folding is often a kinetically inefficient reaction, so that a relatively large proportion of proteins end up in misfolded conformations (Borgia et al., 2015; Mayor et al., 2000). As a result, many proteins require the assistance of chaperones, i.e. specialized proteins that avoid or revert misfolding, to attain their native conformation (Hartl et al., 2011). The main reason for protein misfolding is the existence of conformational frustration, i.e. segments of the primary polypeptide chain that favor non-native conformations (Onuchic et al., 1996). A major source of conformational frustration in proteins is the presence of sequence segments called aggregation-prone regions (APRs) (Ganesan et al., 2016). These segments display a preference for self-association by β-strand interaction, thereby hampering the formation of native interactions. Several chaperone families can recognize such linear APRs, thereby neutralizing their ability to misfold and self-assemble (Rousseau et al., 2006).
Even though the vast majority of protein domains (>95%) possess at least one and often several APRs (De Baets et al., 2014; Ganesan et al., 2016), proteins within a proteome display very differing chaperone requirements and/or dependencies (Fujiwara et al., 2010; Hartl et al., 2011; Niwa et al., 2012). Furthermore, during physiological stress, the ability of different proteins to be rescued by chaperones is also variable. The question as to what protein-specific or cell-context-dependent factors determine chaperone dependency is still poorly understood. A deeper insight into chaperone triage however is of fundamental importance, as protein homeostasis (i.e. protein transcription, translation, folding and degradation) is the major energy consumer of the cell and loss of protein homeostasis is associated with diverse pathologies (Chiti and Dobson, 2017; Sweeney et al., 2017). In addition, protein homeostasis is also limiting as even small physiological stresses on protein homeostasis readily result in a decrease in cellular fitness.
The Escherichia coli protein homeostasis network has three main chaperone systems that help in the folding/disaggregation of polypeptides. These are the Trigger Factor (TF), DnaK/DnaJ and GroEL/GroES systems (Mogk et al., 2011). TF is the first chaperone that interacts with a nascent chain, while it is still attached to the ribosome exit tunnel (Ferbitz et al., 2004). DnaK/DnaJ and GroEL/GroES act downstream of TF (Tyedmers et al., 2010). DnaK/DnaJ and TF share their substrates and one can compensate the absence of the other (Deuerling et al., 2003). A combined deletion of both is lethal above 30°C of growth (Deuerling et al., 1999). Both TF and DnaK/DnaJ bind to hydrophobic sequence segments that are exposed in the denatured, but much less in the native state of a protein, and DnaK in particular shows strong binding when a positively charged amino acid occurs near to the hydrophobic segment (Mogk et al., 1999; Rudiger et al., 2001). Unlike TF and DnaK/DnaJ, GroEL/GroES is the only essential chaperone in E.coli at all growth conditions (Houry et al., 1999). Apart from binding to a sequence segment to promote folding as seen in TF and DnaK/DnaJ, GroEL/GroES offers a microenvironment in its barrel-shaped structure where the full polypeptide can undergo undisturbed folding from rest of the cellular environment (Ellis, 1996). Among these chaperones, the DnaK/DnaJ system in particular is involved in disaggregation of aggregated proteins (Ben-Zvi and Goloubinoff, 2001).
Until very recently it was mostly believed that chaperone dependence (at least of cytosolic proteins) was mainly determined by intrinsic protein factors relating to protein solubility, stability and foldability (Calloni et al., 2012; Niwa et al., 2009; Tartaglia et al., 2010). The main underlying assumption is that there are ‘good’ proteins that are mostly functioning in a chaperone-independent manner and ‘bad’ proteins that require some and sometimes extensive chaperone assistance to fold to their native conformation. This view has recently found additional support with the work of Taguchi and colleagues showing that the in vitro translation of the entire E.coli proteome results in a bimodal distribution constituted of soluble and insoluble proteins (Niwa et al., 2009). Dobson and colleagues have also demonstrated that protein abundance and solubility correlate and have argued that protein solubility has co-evolved to satisfy physiological protein functional requirements suggesting that not only protein folding is encoded in the protein sequence but also the determinants for protein solubility and thus protein abundance (Tartaglia et al., 2009; Tartaglia and Vendruscolo, 2009,, 2010). As a result, this model suggests that the most abundant proteins will also be the best proteins in terms of foldability and solubility and thus those that will be less dependent on chaperones for their folding. This interpretation appears to make sense from a point of view of the cellular energy economy as chaperones could then concentrate their activity on more ‘difficult’ but less abundant proteins, and this both under normal physiological conditions as well as under stress. However, other lines of research suggest that this protein-centric view might not be entirely satisfactory. Indeed, it is now clear that protein translation and protein folding rates are of the same order of magnitude and that as a result both processes are often coupled (Ahmed et al., 2018; O’Brien et al., 2014a; Pechmann and Frydman, 2013; Shiber et al., 2018). Thus, co-translational folding could follow very different folding kinetics than the same protein folding post-translationally (O’Brien et al., 2014a). This principle has been confirmed by forced vectorial protein unfolding by atomic force microscopy showing different unfolding kinetics than free unfolding in solution (Fowler et al., 2002). In addition, it is now also very clear that both average translation rates between different proteins and local translation rates within a protein can vary substantially which again is expected to dramatically affect folding kinetics (Doring et al., 2017; Oh et al., 2011). It is also becoming increasingly clear that evolution uses synonymous mutations between rare and abundant codons to modulate protein translation rates and thereby mechanisms of protein folding in vivo (Pechmann and Frydman, 2013). The question therefore remains how protein-specific factors and cellular context interface with chaperone machinery and determine cellular proteostasis.
2 Materials and methods
The E.coli K12 reference proteome (4305 proteins) and the amino acid sequences of the proteins therein were obtained from UniProt (UniProt, 2008) (proteome ID: UP000000625). Chaperone dependency classifications were determined as outlined in detail in Section 3 and mapped to the reference proteome. The dataset was then expanded with a set of experimentally determined, transcriptome- and proteome-wide features: protein solubility and cell-free expression yield were obtained from the in vitro translation analyses of Niwa et al. (2009); intracellular protein abundance was acquired from the mass-spectrometry-based integrated E.coli dataset from the PaxDb database (Berman, et al. 2000) and from ribosome-profiling data obtained by Li et al. (2014); the latter study also provided data on mRNA abundance and translational efficiency; genome-wide transcriptomic microarray analyses by Esquerre et al. (2016) provided a second mRNA abundance scale, as well as mRNA half-life measurements. Protein melting temperatures (Tm) were obtained from limited proteolysis and mass spectrometry analyses performed by Leuenberger et al. (2017).
To assess nucleotide sequence characteristics, sequences were obtained from the European Nucleotide Archive (Harrison et al., 2019). Protein structures for the calculation of Contact Order (CO) (Plaxco et al., 1998) were retrieved from the Protein Data Bank (PDB) (Berman et al., 2000). When available, the optimal resolution structure with a coverage of at least 40% was retrieved for each protein. Based on nucleotide sequence, primary amino acid sequence and protein structures, the feature-space was expanded using a combination of database cross-references, simple calculations and advanced bio-informatics tools: aggregation propensity and APRs were identified using the TANGO algorithm (Fernandez-Escamilla et al., 2004); The WALTZ algorithm was employed to predict amylogenic regions (Maurer-Stroh et al., 2010); α-helix propensity in the unfolded state was calculated using the thermodynamics algorithm AGADIR (Munoz and Serrano, 1997). Intrinsic disorder calculations were performed using IUPred (Dosztanyi et al., 2005); the EFoldMine method (Raimondi et al., 2017) was used to determine for each protein the percentage of residues predicted to be capable of initiating protein folding. To this end, residues were classified as being part of a foldon if their EFoldMine early folding score exceeds 1.63; GRAVY (grand average of hydropathy) scores were determined by calculating the average hydropathy per protein using the method developed by Kyte and Doolittle (1982); average decoding times were calculated based on decoding time scales devised by Dana and Tuller (2014); isoelectric point values were obtained as the average of different scales using the standalone version of the Isoelectric Point Calculator (Kozlowski, 2016); Codon Adaptation Index (CAI) was calculated using the CodonW software (http://codonw.sourceforge.net//culong.html). Structural classification and protein topology were mapped from the SCOPe (Chandonia et al., 2017) and SUPERFAMILY (Pandurangan et al., 2019) databases. Secondary structure content was calculated from UniProt secondary structure annotations based on a consensus between PDB structures; relative CO (Plaxco et al., 1998) was calculated from the PDB structures by determining the average sequence distance between amino acids that form native contacts, divided by protein length.
Extensive methods are available in supplementary Materials.
3 Results
3.1 An inclusive classification of chaperone substrates
To construct a comprehensive multi-omics dataset on chaperone-substrate interactions and/or dependencies in E.coli we compiled and cross-referenced interaction information of three main bacterial chaperone systems, i.e. TF, DnaK/DnaJ and GroEL/GroES systems from 13 published large-scale experiments (Arifuzzaman et al., 2006; Calloni et al., 2012; Chapman et al., 2006; Deuerling et al., 2003; Fan et al., 2016,, 2017; Fujiwara et al., 2010; Houry et al., 1999; Kerner et al., 2005; Martinez-Hackert and Hendrickson, 2009; Mogk et al., 1999; Niwa et al., 2012, 2016), which can be divided into several categories (Supplementary Fig S1A) based on the detection method. Direct methods aimed at identifying protein–chaperone interactions which was achieved by chaperone co-immunoprecipitation (Deuerling et al., 2003; Houry et al., 1999; Mogk et al., 1999) or his-tag purification (Arifuzzaman et al., 2006; Calloni et al., 2012; Kerner et al., 2005; Martinez-Hackert and Hendrickson, 2009), followed by chaperone client identification by proteomics methods. Indirect methods aimed at identifying chaperone dependencies of protein abundance (Calloni et al., 2012), solubility (Fujiwara et al., 2010; Niwa et al., 2012, 2016), degradation (Calloni et al., 2012) or aggregation (Calloni et al., 2012; Chapman et al., 2006; Deuerling et al., 2003; Martinez-Hackert and Hendrickson, 2009; Mogk et al., 1999) or mRNA profiling (Fan et al., 2016,, 2017) by comparing wild-type E.coli lysates with chaperone deletion/depleted strains. Finally, the in cellulo approaches are complemented with in vitro protein solubility determination using cell-free E.coli translation systems complemented or not with specific chaperones (Niwa et al., 2012, 2016).
These various approaches all have their advantages as well as their specific technical limitations. Direct methods for instance have the advantage of directly monitoring chaperone-client interactions (Supplementary Fig S1A). On the downside, it is hard to distinguish direct from indirect interactions by these methods, that are also biased toward identifying stable interactions while being less suited to detect transient chaperone–client interactions. Indirect methods will encompass the effect of transient chaperone interactions but observed effects have an even larger potential for being indirect or affected by convoluted proteostatic adaptations to chaperone deletion/depletion. Finally, the in vitro translation system, while being the most reductionist system, is probably not fully conservative of cellular protein translation (e.g. in terms of tRNA and amino acid concentrations), which could affect translation rates and therefore protein solubility.
Given this variety in methodologies, it is not surprising that these different studies yield only poor overlap (Supplementary Fig. S1B). Yet it could be argued that they all reveal relevant information on the chaperone dependencies of the E.coli proteome. For this reason, we here decided to merge all the data from these different studies in one meta-dataset and evaluate the union of all the chaperone substrates, rather than to focus on each method separately. This approach has the advantage of creating a more statistically powerful dataset to explore determinants of chaperone dependence. Furthermore, rather than figuring out which study is physiologically the most relevant we considered that creating a meta-dataset would be more alike amalgamating chaperone requirements under a broad range of environmental/experimental conditions. As a result, our meta-dataset not only yields a larger dataset, it also provides a larger window of variation for proteostatic parameters, although it is of course unclear what those are exactly. We here investigated whether such an approach might therefore allow to reveal broader chaperone client properties that are not necessarily apparent in a single experimental setting and/or with fewer data points.
For the large dataset, we merged the data of the 13 aforementioned studies and used the EcoCyc annotation (Keseler et al., 2017) of each protein to focus on cytosolic proteins, and excluded known chaperones, proteases and ribosomal proteins and protein translocation-related chaperones. This left a total of 2198 cytosolic proteins, annotated for their chaperone dependencies (Supplementary Fig. S1C). We classified this dataset in three different manners, which were subsequently analyzed in parallel for various protein-specific and cell-dependent proteostatic dependencies. First, we used a binary classification of proteins as chaperone clients (1617 proteins amounting to 75% of the dataset) versus chaperone-independent proteins (535) to provide a broad picture of the strongest trends differentiating chaperone–clients from chaperone-independent proteins. Second, we also classified our dataset according to whether they were DnaK- (1376), GroEL- (1125) or TF-dependent (993) or chaperone-independent (535). This classification is by definition redundant as many proteins (1217 out of 1617) are dependent of more than one chaperone. The aim of this classification is to evaluate whether chaperone-specific proteostatic parameters still emerge, despite the high redundancy in chaperone function. Finally, having a large enough dataset, we also investigated multi-chaperone dependencies of protein clients by defining eight main chaperone fluxes or pathways (Supplementary Figs S1D and E and S2). These include (i) proteins showing no chaperone dependence in any of the experiments (N), (ii) TF-only clients (T), (iii) DnaK/DnaJ-only clients (K), (iv) GroEL/GroES-only clients (G), (v) both TF and DnaK/DnaJ clients (TK), (vi) both TF and GroEL/GroES clients (TG), (vii) both DnaK/DnaJ and GroEL/GroES clients (KG), (viii) proteins depending on all three systems (TKG). Finally, in a ninth category we grouped proteins for which chaperone dependence was identified but for which the effects were paradoxical, e.g. whereby chaperone knock-down results in an increase in protein abundance. According to this classification, among the 1617 chaperone clients ∼25% are client of a single chaperone, ∼35% of two chaperones and ∼41% are clients of all three chaperone systems (Supplementary Fig. S1D and E and Fig. 1). Overall, this meta-dataset demonstrates that over the broad range of conditions of these various experimental studies, ∼75% of the cytosolic E.coli proteome displays some degree of chaperone dependence, in at least one of the conditions tested. At the same time, the low overlap between the chaperone client repertoires determined by different methods suggests many proteins are conditional clients. The poor overlap could also result from technical limitations affecting sensitivity of detection as well as strain-specific differences. Last, but not least, only a small subset of proteins shows consistent independence from chaperones.
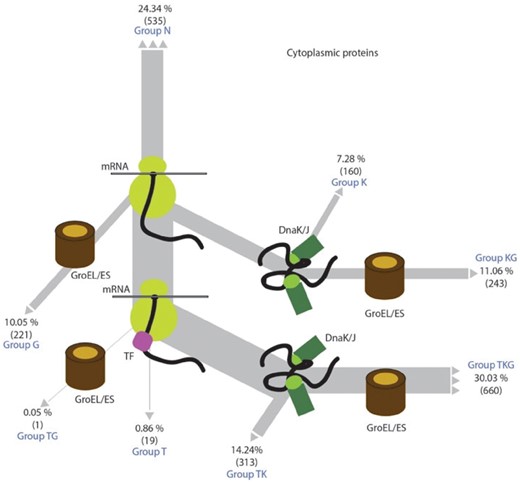
Graphical summary of the routing of cytoplasmic proteins to different chaperones. Different chaperone fluxes and their corresponding classifications are indicated, as well as the percentage of proteins that were identified by our analysis to follow a specific flux
3.2 Chaperone clients have different unfolded state structural properties
To assess whether protein-intrinsic biophysical and/or structural protein parameters determine chaperone dependency we analyzed the meta-dataset introduced above by comparing the statistical distribution of these parameters. Since a brief inspection of the proteome-wide distributions of these parameters frequently revealed bimodal and more complex distributions, we employed the non-parametric Kruskal–Wallis test, followed by post-hoc pairwise Wilcoxon testing with Bonferroni correction for multiple testing. As a simple but robust correction for the additional multiple testing dimension coming from evaluating the large range of properties in our database, we only considered as significant P-values below 10–4. A first thing that stands out is that chaperone clients tend to be highly abundant (Supplementary Fig. S2A), which on the one hand may be expected from a biophysical point of view since protein aggregation is concentration-dependent, but it appears to be in contradiction to the widely held belief that abundant proteins have evolved to fold independently from chaperones for reasons of energy-efficiency (Santra et al., 2017). Second, we confirmed the basic observation that chaperone clients have a significantly lower solubility than chaperone-independent proteins (Supplementary Fig. S2B, P-value = 10–28), although there is a clear biphasic distribution in both groups, meaning that, perhaps counterintuitively, there also appear to be highly soluble chaperone clients. The enrichment of proteins of lower solubility appears to be confirmed by significantly higher total aggregation propensity as predicted by the TANGO algorithm (Fernandez-Escamilla et al., 2004) (Supplementary Fig. S2C), and the fact that chaperone clients have an average isoelectric point that is close to neutral pH, which is known to result in a low colloidal stability (Supplementary Fig. S2D). However, the higher total aggregation propensity seems to be driven mainly by the length of the polypeptides, rather than their intrinsic sequence features, as chaperone clients are strongly biased toward large proteins (Fig. 3E and F, length P-value = 10–53, mass P-value = 10–54). Indeed, there is no difference in the length-normalized intrinsic aggregation propensity between chaperone clients and chaperone-independent proteins (Supplementary Fig. S2G), or in the density of APRs detected by this method (Supplementary Fig. S2H). Similar conclusions were reached with the alternative prediction method WALTZ (Supplementary Fig. S2I) or using a simple proxy like the hydrophobicity of the sequence as calculated by the GRAVY index (Supplementary Fig. S2J). The lower solubility of larger proteins therefore seems to stem from the size-associated accumulation of more aggregation-prone sequences rather than from a higher density of APRs.
When comparing other parameters describing differences between the native structures of chaperone clients and chaperone-independent proteins, we found no difference in the thermodynamic stability as estimated from their Tm (Supplementary Fig. S3A), no difference in their native α-helix (Supplementary Fig. S3B), or β-sheet content (Supplementary Fig. S3C), nor in the level of intrinsically disordered regions predicted by IUPred (Dosztanyi et al., 2005) (Supplementary Fig. S3D). However, the relative CO, a term describing the topological complexity of a protein’s structure, is lower for chaperone binders than spontaneous folders (Supplementary Fig. S3E, P-value = 10–12). This is surprising since high relative CO was shown to correspond to slower spontaneous folding in vitro (Dinner and Karplus, 2001), suggesting that the rate-limiting steps in vitro and in vivo differ significantly. However, the frequency of sequences predicted to be capable of initiating protein folding is significantly lower in chaperone-interacting proteins (Supplementary Fig. S3F, P-value = 10–12). This was predicted using the EFoldMine method, based on early folding data from hydrogen–deuterium exchange from NMR pulse-labeling experiments (Raimondi et al., 2017). This coincides with a higher propensity for α-helix in the unfolded state as predicted by the statistical thermodynamics method AGADIR (Munoz and Serrano, 1997) (Supplementary Fig. S3G, P-value = 10–27), which was shown to be able to slow down protein folding by stabilizing non-native local structure in the unfolded state (Viguera et al., 1995) as well as inhibit β-aggregation (Fernandez-Escamilla et al., 2004). This suggests that in vivo the critical steps for folding occur already in the unfolded state: chaperone clients both display lack of early foldons and a level of structural frustration slowing down aggregation kinetics, that conspire to slow down their spontaneous folding, driving them toward chaperones (Bandyopadhyay et al., 2017).
Despite these findings none of the differences in these simple structural or biophysical parameters directly affecting protein stability, folding and aggregation are sufficiently large to allow segregating chaperone clients from chaperone-independent proteins from single factors. Reanalysis of these parameters considering either dependence on a specific chaperone (four categories) or dependence on multi-chaperone fluxes (eight categories) did not yield additional resolution.
3.3 TF/DnaK-dependent fluxes handle abundant proteins with high translation rates
As we cannot detect simple structural determinants of chaperone dependence the question therefore arises whether the specific physiological context in which folding occurs is a bigger determinant of chaperone dependence. Contrary to in vitro equilibrium conditions, protein folding and quaternary protein structure assembly largely occurs co-translationally (Khushoo et al., 2011; Nicola et al., 1999; O’Brien et al., 2014a; Pechmann and Frydman, 2013). As the timescales of protein translation kinetics and protein folding kinetics are of the same order of magnitude, they can interfere which each other so that translation can affect protein folding efficiency (O’Brien et al., 2014a; Pechmann and Frydman, 2013) and vice versa (O’Brien et al., 2014a,b). As previously mentioned, we find a strong enrichment of highly abundant proteins in chaperone clients. When classifying by individual chaperones (Fig. 2A and B) we find this enrichment of abundant proteins in all three categories (TF P-value = 10−50, DnaK P-value = 10−32, GroEL P-value = 10−18). However, classifying by multi-chaperone fluxes (Fig. 2C) we find that the enrichment of abundant proteins specifically occurs in the TF/DnaK-dependent pathways, but not GroEL-only clients. The same observation is evident from mRNA abundance: High mRNA abundance is enriched in chaperone clients (Fig. 2D and E, P-value = 10−40), but here as well this enrichment is strongly associated with TF/DnaK-dependent fluxes (P-values ranging 10−35 to 10−67, Fig. 2F), but it is absent from DnaK-only or GroEL-only dependent fluxes (Fig. 2F). Interestingly, in the category showing paradoxical response to chaperone deletion, i.e. an increase in abundance of mRNA or protein, both protein and mRNA abundance are also increased, which is consistent with the role of TF as a molecular brake of translation-associated folding (Merz et al., 2008). However, next to abundant proteins, chaperone clients are enriched in fast translating proteins (Fig. 2G and H), which again is strongly associated with TF-dependent chaperone fluxes (P-values ranging 10−20 to 10−24, Fig. 2I) while GroEL-only clients show a modest enrichment in slowly translating proteins (P-value 10−3, Fig. 2I). Interestingly, we find that low mRNA half-life is also strongly enriched in chaperone clients (Fig. 2J and K), again along the TF/DnaK axis (Fig. 2L, P-values ranging 10−26 to 10−42). This suggests that abundant and highly expressed proteins require high mRNA turnover, possibly to maintain low-density polysomes and avoid interference between nearby nascent chains on polysomes that could result in jamming. Finally, these observations are confirmed by the fact that chaperone clients also display a significant codon usage bias, as calculated from the CAI (Lee et al., 2010), a score that increases with the proportion of translationally optimal codons in a gene (Fig. 2M and N). This is again attributable to TF/DnaK-dependent chaperone fluxes (P-values ranging 10−19 to 10−22, Fig. 2O). It is clear that all these parameters describing translation and abundance are intercorrelated, but since there is no simple correspondence between the values, it makes sense to analyze each feature separately. These findings suggest that TF/DnaK-dependent fluxes handle highly abundant and therefore often fast translated proteins, which is associated with both a high codon bias and fast mRNA turnover.
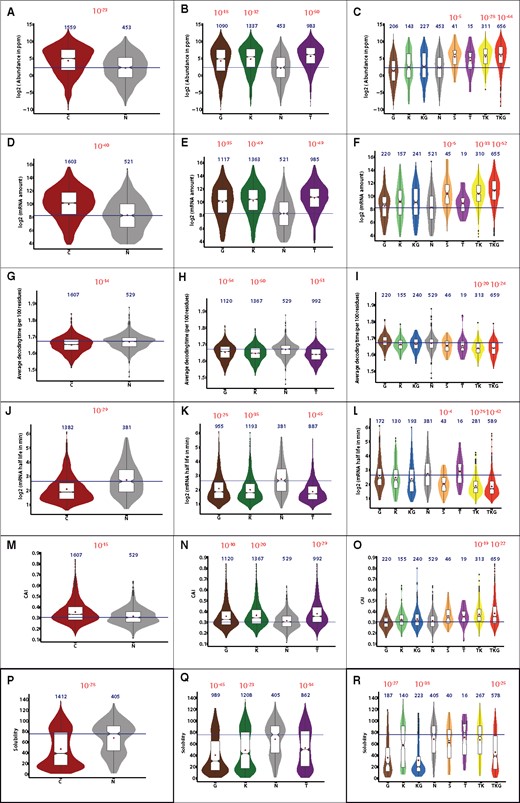
Distribution of cell-specific parameters and a comparison between different grouping strategies. Distributions are shown through a combination of boxplots (in white) and violin plots (colored) as described in Supplementary Figure S2. Plots are arranged per parameter (one per row) with one grouping strategy per column. The first column shows the simple grouping into chaperone-dependent folders (C) and spontaneous folders (N). The second column shows the grouping of proteins into clients of each individual chaperone system (G for GroEL, K for DnaK, T for Trigger Factor and N for spontaneous folders). This grouping is inherently redundant as one protein may be client to several chaperone systems. The third column depicts grouping into six different chaperone fluxes (G: GroEL only, K: DnaK only, KG: DnaK and GroEL, T: Trigger Factor, TK: Trigger Factor and DnaK, TKG: Trigger Factor, DnaK and GroEL) as well as spontaneous folders (N) and a group of proteins with paradoxical classification (S). (A–C) Abundance in p.p.m. determined by mass spectrometry (Wang et al., 2015). (D–F) mRNA abundance (Li et al., 2014). (G–I) Average decoding time (Dana and Tuller, 2014), (J–L) mRNA half-life (Esquerre et al., 2016), (M–O) CAI (Lee et al., 2010). (P–R) Solubility during cell-free translation (Niwa et al., 2009)
3.4 Translation rates modulates the GroEL dependence of topologically complex folds
As previously mentioned chaperone clients are significantly enriched in insoluble proteins (Supplementary Fig. S2B) as well as abundant proteins (Fig. 2A–C). Dissecting multi-chaperone dependencies however shows that these two properties distribute over different chaperone fluxes. TF/DnaK-dependent fluxes are enriched in abundant but more soluble proteins (Fig. 2P–R), while GroEL/DnaK-dependent fluxes are enriched in less soluble but non-abundant proteins (Fig. 2R). Only clients that are dependent on all three chaperone systems display both high abundance and low solubility (Fig. 2C and 5R). Interestingly, the DnaK-only group (160 proteins) does not display any enrichment toward solubility, nor abundance (Fig 2C and 2R), confirming the status of DnaK as a universal chaperone which can support the folding of TF-dependent abundant and fast translating proteins, as well as GroEL-dependent poorly soluble proteins. As reported previously by other studies, the GroEL-dependence of its clients could partially be explained by the fact that they are enriched in proteins with complex, difficult-to-fold topologies that require GroEL’s ‘Anfinsen cage’ to be able to fold. To analyze this, we performed an enrichment analysis of superfamilies of the SCOP classification (Andreeva et al., 2004) in our different fluxes over proteome average. The superfamily level groups protein families with a similar fold but no detectable evolutionary relationship, whereas within a family the proteins are also evolutionarily related. We represented these results in so-called ‘volcano plots’, which show the fold enrichment of each SCOP superfamily versus the P-value of each enrichment calculated by the Fisher-exact test, which allows to identify significantly enriched folds in each chaperone flux (Supplementary Fig. S4A–G). To perform this analysis we employed the Superfamily database (Pandurangan et al., 2019) in which SCOP annotations of entire proteomes can be obtained, using a method based on a Hidden Markov Model that requires only the sequence of each query protein (Gough et al., 2001). In agreement with previous reports, we only found significantly enriched topologies (superfamilies) in the different fluxes involving GroEL/ES (Supplementary Fig. S4A and E–G) (Houry et al., 1999) and not those depending on DnaK/J or TF (Supplementary Fig. S4B, C, and E). Also, well within expectations, most of the enriched superfamilies stem from the C class in SCOP, which is the topologically most complex category involving elements of α- and β-structure that occur interspersed in the primary structure, although some exceptions confirm this rule (Supplementary Table S1). This confirms the earlier proposed notions that GroEL specializes in hard-to-fold topologies, but since the fold enrichments are often fairly low, topogical selection is certainly not absolute and for each enriched superfamily, there are also members that fold spontaneously. To resolve this, we had a closer look at the previously reported TIM barrel family, which is often cited as a topology in relation to GroEL clients (Houry et al., 1999) as this is one of the most abundant folds in protein space, although it falls just outside the enrichment criterion in our analysis (log fold enrichment = 1.47, P-value = 5.10−5). Even though the fold is conserved, its member proteins exhibit a high functional diversity [33 superfamilies in SUPERFAMILY database (Pandurangan et al., 2019)] and often lack sequence similarities even between proteins in the same functional class, making their evolutionary history hard to track. Several members have been shown to interact with GroEL/ES (Fujiwara et al., 2010; Hirtreiter et al., 2009; Houry et al., 1999; Kerner et al., 2005), but also here this dependence is not universal throughout the superfamily as many TIM barrel proteins have been reported among the most highly soluble proteins when expressed in vitro in a cell-free translation system in the absence of any chaperones (Fujiwara et al., 2010; Niwa et al., 2016).
For example, enolase, an abundant enzyme of E.coli with with two folds, a TIM barrel fold (c.1) and an enolase N-terminal domain-like fold (d.54), shows spontaneous refolding upon dilution from denaturant and regains more than half of its activity (∼55%) under standard conditions (Kerner et al., 2005). The solubility of enolase is 101% in the cell-free (and chaperone-free) translation system (Niwa et al., 2009). Although recently two different studies suggested that folding rate and local frustration, similar to our findings above, may play a role in chaperonin dependency (Bandyopadhyay et al., 2017; Georgescauld et al., 2014), it remains unclear at present what makes some TIM barrels obligate GroEL substrates and some spontaneous folders. Since we found the GroEL/ES axis has an enrichment of slow translated proteins (Fig. 2I), we reanalyzed TIM barrel proteins in E.coli to determine how well this property separates obligate GroEL/ES and spontaneously folding TIM barrel proteins in E.coli. To avoid noise and to obtain precise information we restricted our analysis to an experimentally validated set of TIM barrels with known GroEL/ES dependency. The list of proteins was obtained from four different studies (Fujiwara et al., 2010; Kerner et al., 2005; Niwa et al., 2012, 2016) and consists of 30 cytoplasmic TIM barrel proteins that are obligate GroEL/ES clients and 28 TIM barrel proteins that can fold independently of GroEL/ES. The solubility obtained with cell-free translation of these two groups is fundamentally different (Supplementary Fig. S5A), confirming their classification into obligate chaperone substrates and spontaneous folders. The CAI already shows a difference between these two groups at the gene level (Supplementary Fig. S5B), and when we evaluated the difference in decoding time (Supplementary Fig. S5C) or translational efficiency (Supplementary Fig. S5D), we observed the same trend as for GroEL substrates overall, that obligate GroEL client TIM barrels show longer decoding times and lower translational efficiency.
3.5 Validation through a reanalysis of cross-sectional data
As an alternative approach, we repeated the key findings but restricting the analysis to chaperone substrates that are consistent between studies, which yields a much smaller but higher confidence dataset. To this end, from the Supplementary Figure 1, B1, B2 and B3, the proteins in the outer most layers are removed. These are the proteins that were detected only once in a particular study and it could be argued that they cannot be reliably classified to any category, so removal of these protein from the chaperone clients ensures a reduction in noise additional to the above grouping strategies (especially the nine groups). After removing the proteins in the outer layers, which in–total constitute 1453 proteins (or we put all of them to a single group), the chaperone substrates dropped from 1617 to only 187, divided over the categories as follows; 72 (TKG), 19 (TK), KG (23), K (15), G (58), T (0), TG (0). We verified that for the main findings we found the same conclusions than for the more inclusive approach: mass (Supplementary Fig. S6A), solubility (Supplementary Fig. S6B), abundance (Supplementary Fig. S6C), decoding time (Supplementary Fig. S6D) and CAI (Supplementary Fig. S6E).
3.6 Manipulating translation rates to modulate chaperone dependencies
To observe the effects of translation rate on chaperone dependency in an experimental setting, we employed synonymous mutations to design three variants of GFP with identical amino acid sequences, yet varying translational efficiencies. Apart from a wild-type version, we designed a variant with low translational efficiency in E.coli (‘slow’ variant), and high translational efficiency (‘fast’ variant). We designed these variants using the codon decoding time scales devised by Tuller and colleagues (Dana and Tuller, 2014), and replaced every codon with either its fastest or slowest translating counterpart, for the fast and slow variants, respectively (Fig. 3A). To assess chaperone dependencies, these protein variants were expressed in an in vitro translation system (NEB PURExpress®) with or without the addition of either DnaK mix, which consists of mixture of DnaK, DnaJ and GrpE or GroE mix, which contains both GroEL and GroES. The use of a reconstituted in vitro translation system offers the advantage of being completely devoid of proteostatic machinery components such as molecular chaperones and proteases, and therefore allows for a very clean interpretation of the effects of the addition of individual chaperones. To determine the effects of translation rate on solubility, proteins were expressed for 1 h, after which protein solubility was assessed (Fig. 3B and C). Clearly, increasing translation rate decreases soluble protein expression under control conditions. DnaK addition does not significantly increase solubility for the wild-type and slow variants, but does do so for the fast variant, confirming our observation that abundant proteins with high translation rates rely more strongly on DnaK. Addition of GroE mix however, does not significantly increase solubility of any of the constructs.
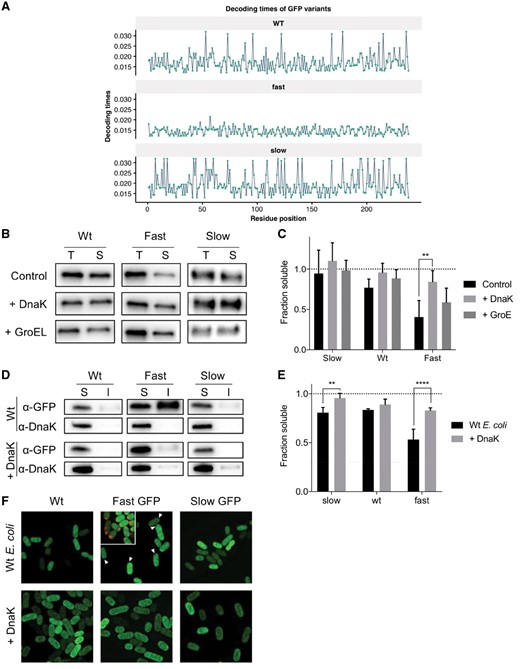
Experimental analysis of codon usage in the chaperone dependence of GFP. (A) Decoding times of our GFP variants. Decoding time is shown per residue position, with each panel representing one of the variants. (B) Representative western blot of the solubility analysis of GFP variants upon cell-free expression. Bands show Total (T) and Soluble (S) fractions after centrifugation at 21 000g for 30 min. ‘+ Dnak’ and ‘+ GroEL’ indicate addition of either DnaK mix—containing DnaK, DnaJ and GrpE—or GroE mix—containing GroEL and GroES—respectively. (C) Bar plots showing mean solubility as determined through quantification of western blot bands in (B), whiskers indicate standard deviation (n = 5). Statistical significance was determined through two-way ANOVA followed by Tukey’s post-hoc test (‘**’ indicates P-value <0.0021). (D) Representative western blot of the solubility analysis of GFP variants upon overexpression in E.coli K12. Bands show Soluble (S) and Insoluble (I) fractions after centrifugation at 17 100g for 15 min. ‘+ Dnak’ indicates co-expression of pKJE7, encoding DnaK, DnaJ and GrpE. (E) Bar plots showing mean solubility as determined through quantification of western blot bands in (B), whiskers indicate standard deviation (n = 4). Statistical significance was determined through two-way ANOVA followed by Bonferroni’s post-hoc test (‘**’ indicates P-value < 0.0021, ‘****’ indicates P-value < 0.0001). (F) Structured Illumination Microscopy images of E.coli after 3 h of expression of one of the GFP variants. GFP fluorescence is show in green. ‘+ Dnak’ indicates co-expression of pKJE7, encoding DnaK, DnaJ and GrpE. White arrows indicate the position of non-fluorescent inclusion bodies. The inset in the top center panel shows an overlay of intrinsic GFP fluorescence in green, and ReASH-EDT2 labeled GFP fluorescence in red
To assess in cellulo effects of altered translation kinetics, the same set of constructs was overexpressed in E.coli K12, and solubility assessed. Wild-type and slow GFP produce mostly soluble protein, resulting in diffuse fluorescence throughout cells (Fig. 3D–F). Increasing translation rate however, renders half of the GFP produced insoluble and clustered into non-fluorescent inclusion bodies. We verified the presence of GFP in these inclusions through a tetracysteine tag, which was stained using ReASH-EDT (ThermoFisher; Fig. 3F, inset). Co-overexpression of DnaK with fast GFP rescues the protein from going insoluble, and results in completely diffuse staining, in agreement with the in vitro results.
Together, these data show that increasing the translation rate of GFP changes the dependencies of the protein, causing it to become more dependent on DnaK for its solubility. In other words, merely increasing the translation rate without affecting intrinsic properties of the protein reroutes GFP from a mostly DnaK-independent flux to a DnaK-dependent flux, showing that translation rate is in fact a strong determinant for chaperone dependency, independent of protein-intrinsic characteristics.
4 Discussion
Chaperone-interaction and -dependence data have previously been generated for E.coli as well as for other organisms using a multitude of different approaches. Overall these studies suffer from a lack of overlap in chaperone clients which in turn made it difficult to identify general protein structural or proteostatic properties determining chaperone dependencies. Rather than trying to figure out which experimental approach is most effective in uncovering physiologically meaningful chaperone clients we here reasoned that all these studies should be considered equally as they all map real chaperone dependencies. Rather than only introducing noise, the addition of these different experimental results then is the equivalent of building a dataset that explores the proteostatic landscape under a wider variety of conditions. The immediate consequence of this approach is that about 75% of the 2198 cytosolic E.coli proteins considered in this dataset display some degree of chaperone dependence under at least one of the experimental conditions while of course only a very limited set is found to have the same chaperone dependencies in all studies. This added advantage of statistical power also allows to study multi-chaperone dependencies in more depth.
Our meta-analysis confirms that chaperone clients have a significantly lower average solubility than chaperone-independent proteins but also that some proteins are chaperone clients despite being highly soluble. When looking for structural parameters we found not so surprisingly that chaperone client are significantly enriched in larger proteins. However, when normalizing for size we could not find any difference in structural features defining the native state and its stability. Thus, for an average protein domain size there is no significant difference in density of aggregation-nucleating regions, hydrophobicity, secondary structure, thermodynamic stability or intrinsic disorder content between chaperone dependent and independent proteins.
Interestingly however we found significant differences in structural parameters determining folding kinetics. Indeed, chaperone clients are enriched in proteins displaying a lower frequency in early foldons, i.e. polypeptide segments having a high propensity to readily adopt native-like structure in the unfolded state. At the same time chaperone clients display a significantly higher helical tendency in the unfolded state which often is not the native secondary structure and therefore indicative of some degree of structural frustration. Together the lack of early foldons and (non-native) helical structural frustration in chaperone clients suggest less efficient co-translational folding kinetics allowing for more efficient chaperone interactions. Interestingly this contrasts with the maybe counterintuitive observation that chaperone clients are significantly enriched in proteins with low CO in their native structure, a feature which correlates with faster protein folding kinetics under post-translational equilibrium conditions. Together this might reflect the selective co-optimization of chaperone clients for efficient chaperone interactions during translation with an efficient post-translational folding rate when chaperones leave these proteins to their own devices.
Next to intrinsic protein structural parameters we also investigated cell-context-dependent proteostatic parameters of chaperone clients. Contrary to the structural parameters described above we found that proteostatic parameters clustered according to specific (multi)chaperone dependencies. TK-dependent clients are highly enriched in fast translating and abundant proteins but have a solubility distribution that is not significantly different from chaperone-independent proteins. However, G- and GK-dependent clients display very low solubilities and protein abundance but have similar translation rates as chaperone-independent proteins. Finally, only proteins dependent on TKG display low solubilities together with high abundance and/or translation rates. These findings suggest that the proteostatic regulation of the E.coli proteome by TF, DnaK and GroEL is organized along two different but sometimes also overlapping needs: (i) avoiding misfolding and aggregation of abundant and/or fastly translating proteins and (ii) avoiding misfolding and aggregation of low solubility proteins. The specialization of the TF/DnaK axis to protein abundance and fast translation rates as found here is also confirmed by associated proteostatic parameters such as high mRNA levels in conjunction with high mRNA turnover rates (which very likely regulates polysome occupancy) as well as a significant codon usage bias toward fast translating codons.
These findings do not contradict but rather complement previous findings that chaperones and particularly GroEL favors topologically complex folds with low abundance and solubility. Indeed here we confirmed the low solubility and abundance of GroEL clients as well as the enrichment of mixed α/β topologies for GroEL chaperone-dependent clients. In addition however, comparing cell-dependent proteostatic parameters for a set of validated GroEL obligate and GroEL-independent TIM barrels, we found that obligate GroEL TIM barrels not only have a significantly lower solubility but that they also have a lower translation rate as well as a codon bias toward slow translation, suggesting that here as well solubility, translation rate and codon biases are interrelated.
Finally, we recapitulated some of the above finding experimentally by comparing WT GFP with both codon-optimized slow translating and fast translating GFP. While slow translating GFP does not significantly differ from WT GFP due to the fact that WT is already a slow translating protein, the situation is different for fast translating GFP. Both in vitro and in E.coli cells it was found that fast translating GFP was more abundant but also less folding competent, forming nonfunctional inclusion bodies. However, complementing fast GFP with DnaK did efficiently rescue GFP solubility while partial less efficient rescue was also observed by GroEL.
Together our results therefore highlight the dual nature of chaperone regulation in E.coli which is both geared toward difficult to fold insoluble proteins and also to abundant and fast-translating proteins.
Acknowledgements
We thank the following researchers for feedback on methods and calculations: Hideki Taguchi (Tokyo, Japan), Tamir Tuller (Tel Aviv, Israel), Wim Vranken (Brussels, Belgium) and James McInerney (Manchester, UK). Geert Molenberghs (K U LEUVEN, Belgium) for statistical expertise.
Funding
The Switch Laboratory was supported by grants from the European Research Council under the European Union’s Horizon 2020 Framework Programme ERC [647458 (MANGO) to J.S.], the Flanders Institute for Biotechnology (VIB), the University of Leuven (‘Industrieel Onderzoeksfonds’), the Funds for Scientific Research Flanders. (FWO), the Flanders Agency for Innovation by Science and Technology [IWT, SBO 60839] and the Federal Office for Scientific Affairs of Belgium (Belspo), IUAP [P7/16]. R.R. was supported by an Erasmus Mundus fellowship. B.H. was supported by PhD Fellowship from the IWT. Structured Illumination Microscopy was performed at the VIB Bio-imaging Core at KU Leuven.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}