





Abstract
Genome repositories are growing faster than our storage capacities, challenging our ability to store, transmit, process and analyze them. While genomes are not very compressible individually, those repositories usually contain myriads of genomes or genome reads of the same species, thereby creating opportunities for orders-of-magnitude compression by exploiting inter-genome similarities. A useful compression system, however, cannot be only usable for archival, but it must allow direct access to the sequences, ideally in transparent form so that applications do not need to be rewritten.
We present a highly compressed filesystem that specializes in storing large collections of genomes and reads. The system obtains orders-of-magnitude compression by using Relative Lempel-Ziv, which exploits the high similarities between genomes of the same species. The filesystem transparently stores the files in compressed form, intervening the system calls of the applications without the need to modify them. A client/server variant of the system stores the compressed files in a server, while the client’s filesystem transparently retrieves and updates the data from the server. The data between client and server are also transferred in compressed form, which saves an order of magnitude network time.
The C++ source code of our implementation is available for download in https://github.com/vsepulve/relz_fs.
1 Introduction
Since the first human genome was sequenced with the turn of the millennium, the cost of whole-genome sequencing has dropped to a few hundred dollars, becoming a routine activity. The amount of sequenced genomes has been growing faster than Moore’s Law (Sthephens et al., 2015), and threatens to overflow our storage capacity.
Fortunately, those genome collections are highly repetitive, because they consist of many genomes of the same species, differing from each other in small percentages only. It is then possible to sharply compress them with Lempel-Ziv compression programs like p7zip (http://p7zip.sourceforge.net). However, once the collection is compressed, we need to decompress it completely in order to extract a single genome, or to access a short snippet of it. Therefore, this is an efficient long-term archival solution only. There exist other compression mechanisms that exploit repetitiveness almost equally well while supporting direct access to the compressed collection, such as grammar compression (Kieffer and Yang, 2000) and block trees (Belazzougui et al., 2015). None of them, however, allow for efficient updates on the collection, e.g. adding, removing or modifying genomes.
Relative Lempel-Ziv compression (RLZ) (Kuruppu et al., 2010) is a recent technique that adapts particularly well to this scenario. In RLZ, we choose a reference text (which can be, for example, one genome from each involved species), and represent all the other texts as sequences of (long) substrings of the reference. In the case of genome collections, RLZ obtains compression ratios similar to plain Lempel-Ziv compression, while allowing for a much more flexible access to the data: it is possible to efficiently access the compressed file at random positions (Cox et al., 2016; Deorowicz and Grabowski, 2011; Ferrada et al., 2014), and even to modify a genome, or to add and remove whole genomes. There exist even indexing proposals to allow for fast pattern searches on the collection, though they have not been implemented (Do et al., 2014) or do not support updates (Belazzougui et al., 2014; Farruggia et al., 2018).
In this article we go further than designing algorithms for accessing and updating RLZ-compressed collections: we demonstrate that it is feasible to build on RLZ to design a fully-functional compressed filesystem specialized on managing genome collections. Our filesystem uses RLZ to maintain a few references in plain form and all the other genomes in compressed form. It intervenes all the typical system calls to open, close, read, write, etc., so that applications can transparently use the filesystem without need to modify them.
Our filesystem can also be used in client/server form. The server then maintains a large centralized repository of genomes, potentially offering stronger compression. When the intervened filesystem of the client needs to open a file, the file is transferred from the server in compressed form, saving orders of magnitude network transfer time (which is a serious problem when storing large files remotely). The file is then stored locally in compressed form, where it can be accessed and updated. Transfers of new or modified files to the server also proceed in compressed form.
Overall, the compressed filesystem expands the storage capabilities of the client computers by orders of magnitude, saving also orders of magnitude in its own storage space and network transfer time. Our experiments show reductions to 1–3% of the original space and to around 10% of the original network transfer time.
In many cases, however, the sequencing data do not form fully-assembled genomes, but consist of partially assembled contigs or, more frequently, simple reads of a few hundred or thousand bases. Our system can be adapted to store large numbers of such short sequences. Even when a reference genome is unavailable, the system is able to build an artificial reference from the data, which manages to reduce the space to 12% of the original.
2 Materials and methods
2.1 Relative Lempel-Ziv compression
RLZ (Kuruppu et al., 2010) compresses one sequence with respect to another, hopefully similar, sequence . It processes S left-to-right. At each point, where has already been processed, it looks for the longest prefix of that appears somewhere in R. If this longest prefix is , then RLZ outputs the pair , and restarts the process from . A special case occurs if does not appear in R, in which case RLZ outputs . Alternatively, we can append all the alphabet characters to R to ensure this case does not arise. We will assume the latter choice in this article for simplicity.
The output of RLZ is then a sequence of z pairs , so that . When S and R are very similar, S is represented as the concatenation of a small number z of substrings of R (also called factors of R). The process of splitting S into a set of factors of R is called the factorization of S.
2.2 Suffix arrays
An efficient way to implement RLZ compression is via a suffix array (Manber and Myers, 1993). The suffix array of a sequence is an array of integers containing the starting positions of the r suffixes , sorted in increasing lexicographic order. That is, A is a permutation of so that for all , in lexicographic order. It then holds that all the positions where a given string occurs in R are contiguous in A, so this range can be binary searched in time .
The suffix array of R can be used to find the longest prefix of occurring in R. We binary search A for the whole suffix . If we find it in, say, , then we are done and can emit the final pair . If not, the binary search will give us the position such that . We can then form the new phrase with the longest prefix of or that matches a prefix of . Figure 1 illustrates the process.
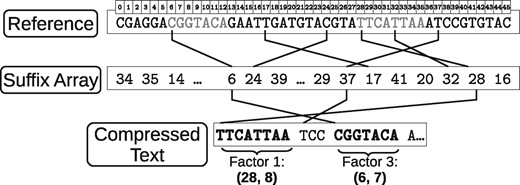
RLZ factorization using a suffix array
When forming a pair of length , we never compare more than symbols, and since the lengths of all the pairs add up to n, we carry out the whole RLZ compression in time . It is possible to carry out this compression more efficiently, for example, using an FM-index (Ferragina and Manzini, 2005), which is also more space-economical. However, we prefer to use a suffix array because we will then be able to use a sampled version of it, which will be orders of magnitude smaller.
3 Implementation
3.1 Sequence compression
We compress a new sequence S with respect to a reference sequence R using RLZ with the help of the suffix array A of R, as described in Section 2. Note that the suffix array A is not needed for decompression; just the reference R and the pairs that represent S suffice.
However, the space of A may pose a significant overhead at compression time, especially when compression is carried out on computers with little available memory. For , A requires four times the size of R. (Moreover, if we pack the symbols of R in 2 bits, then A is 16 times larger.) To reduce the space of A, we use a sampled version of it: we choose a sampling factor s (which is in practice in the tens or hundreds) and preserve only the entries i such that is a multiple of s. This reduces the storage requirements of A by a factor of s, which as said can be one or two orders of magnitude.
The result of the RLZ compression on a sampled suffix array is still a valid compressed file, though it can be larger than the file obtained with the full suffix array: only the factors of R starting at multiples of s can be used to form pairs in S, and thus the longest factor is not chosen in most cases. While sampling yields no guarantee on the degradation that the compression ratio may undergo, it is expected that the pairs are shortened by an amount around s, which should imply only a small impact on compression. Figure 2 illustrates the sampled factorization.
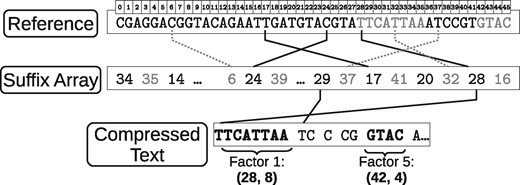
Factorization with a sampled suffix array. Some positions (marked in dotted line and gray) are not available in the array, so other factors are used instead, generating a slightly worse compression
Once the sequence of pairs is determined, the pairs are encoded in a way that uses less space than just using plain integers. We process the positions and the lengths separately with appropriate techniques for their encoding. First we look for , the highest position of the factors from the compressed text segment, and we use bits to encode each consecutive position of each pair in binary form. Since each position uses a fixed number of bits, they can be easily positionally accessed for decompression. It is possible to use other techniques to encode the positions that may use less space [Variable Byte (Williams and Zobel, 1999), Golomb/Rice (Golomb,1966), etc.], but in our tests the gain in space was negligible and the negative effect on decompression times was noticeable. On the other hand, factor lengths are significantly compressed using Golomb codes (Golomb, 1966). That is, we use a power-of-2 parameter M to divide each number N from the input and generate two values that are stored separately: the quotient and the remainder . The quotient q is encoded in unary (using q + 1 bits), and the remainder is encoded in binary form using bits. In our case, we use M = 64. Sampled pointers to the Golomb-compressed file enable efficient direct access to any factor.
The output of the process is then a compressed binary file that encodes the pairs, preceded by a header with metadata with some basic information on the file that was compressed. Figure 3 illustrates the whole process.
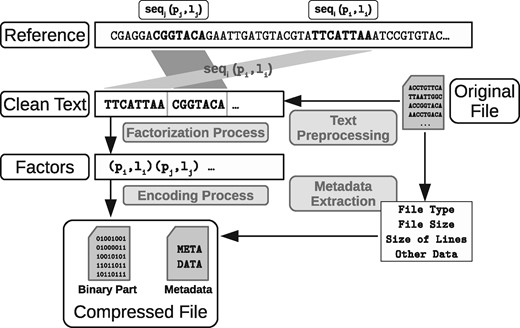
Our complete compression method
3.2 Compression by blocks
While it is possible to access random positions of the RLZ compressed sequence in constant time (Cox et al., 2016; Ferrada et al., 2014), our compressor must efficiently support not only positional access to the files, but also updates and appends. Therefore, a more flexible solution is needed. We (conceptually) split the original files into blocks of fixed size blocksize, which are compressed independently [in the same style of previous work (Deorowicz and Grabowski, 2011)].
This arrangement makes it very easy to identify the blocks involved in positional operations like the system call read(pos, len), which retrieves the text of length len from the file position pos. Except for the last block, which may be smaller, all the other blocks are of length exactly blocksize, so the blocks that need to be decompressed range from to .
Compressing the blocks independently limits the maximum length of the factors obtained by the RLZ factorization, because a factor cannot cross blocks in S. This may worsen the compression ratio of the whole file, but the damage is limited: at least using the full suffix array to find the factors, compressing the blocks independently does not add more than n/blocksize factors in a file of size n.
To use the blocks for efficient access, we must know the position of each block in the final compressed file. At the end of the encoding process, the size in bytes of each block is stored in a header of the binary file. After the header, the bytes of each successive compressed block are written.
Since the different file blocks are compressed independently and the result of the compression of one block does not affect the others, these different compression processes can be performed in parallel.
The compression system uses a pool of compressor threads. At the time of compressing the blocks of a file, the amount of threads to use is determined and the threads are initialized. Then, the segments of the input file corresponding to each block are stored in a work queue with exclusive access. The threads consume these segments in parallel, performing the compression of each block and storing the data in local files of each thread, together with information that will be used by the data consolidation process. When the work queue becomes empty, we have all the text blocks compressed in different files, along with the consolidation data. The compression process ends with the consolidation phase of the blocks and the construction of the final file. In this phase, the metadata is prepared and the headers are written in the file. Finally, the bytes of each block are read in order (recorded by the proper thread in its consolidation data), and they are written in the final file following the header. Figure 4 shows the full compression process.
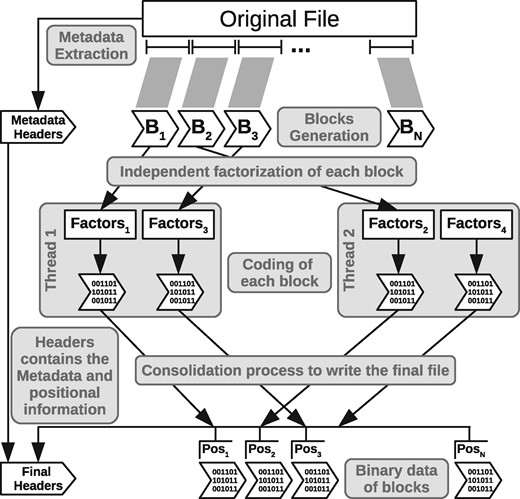
Compression by blocks
As mentioned, the block system facilitates positional reading and writing without the need to decompress the entire file. For reading, we decompress only the blocks that overlap the segment of the file that must be read. For writing, we must implement the operation write(buf, pos, len), which replaces len bytes from file position pos with those from the buffer buf. This may eventually extend the file beyond its current length. We find the blocks that will be modified and decompress them. We then replace their contents from position pos, and recompress them into a temporary file. To complete the process, the blocks of the original file are merged with the new blocks, in binary form, into a new compressed file. Since the compressed files are usually much smaller than their uncompressed versions, the time to copy the blocks already compressed to consolidate the final file is negligible.
In the event that the data to be added to the compressed file exceeds its original size, the process may create additional blocks. In particular, appending new data at the file tail, in practice the most common type of writing, is done very efficiently. Figure 5 illustrates the writing process in our compressed files.
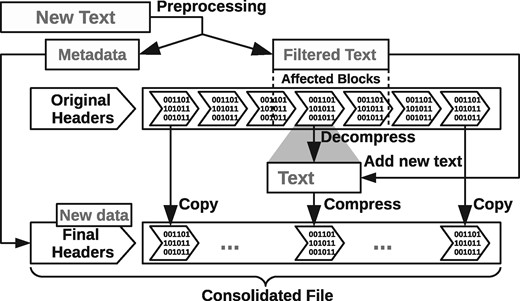
Writing in the compressed file
3.3 File formats
The RLZ compression approach, as described in Sections 3.1 and 3.2, works very well to store genomic sequences. In practice, the file formats used to store those sequences contain additional text, such as different kinds of metadata. Our software handles the FASTA standard format, although it can easily be extended to handle other formats as well. FASTA-formatted files include metadata in lines starting with a special character (‘>’ or ‘;’). Not only the usual RLZ approach is not the most appropriate to deal with metadata, but also those lines could break a potentially long factor of the sequence. To support metadata, we first extract those lines and process them in a stream separate from the sequence. We also divide the metadata text into blocks, in a similar way to the sequence, but we use Lempel-Ziv-Markov chain algorithm (LZMA, https://www.7-zip.org/sdk.html) to compress the blocks.
At the moment of accessing the compressed text by means of a call such as read(pos, len) or write(buf, pos, len), we extract both the text from the sequence and from the metadata, and use positional information stored while separating both streams to combine them in the final decompressed blocks. Both the compressed text from the metadata and the positional information necessary to combine it with the sequence are stored as a part of the header in the final compressed file.
3.4 Reference generation
We have assumed up to now that the files to handle are fully-assembled genomes. Our system can also handle partially-assembled sequences, such as contigs or simple reads from a genome. In cases where a fully-assembled genome is present and can be used as a reference, our scheme does not need any modification. We note, however, that the lengths of the factors will be limited by those of the files to compress, and therefore we cannot expect the same compression ratios when representing, say, reads of a few hundred bases, than when representing whole genomes.
A more challenging scenario arises when we only have a set of short sequences (say, reads) and no reference sequence of the genome they belong to. To handle this case, we provide a simple procedure to generate an artificial reference based on the data: we randomly extract segments of a certain length from the dataset and concatenate them to build the reference. The metadata from the processed files is omitted in this phase, since the reference is only used for the compression of the sequence data. The length of the segments is a parameter of the reference generation program; in practice it should be hundred to thousand bases. The size of the generated reference is also a parameter, and it depends on the amount of data and the availability of resources on the machine holding the reference.
More sophisticated techniques to form a reference exist (Liao et al., 2016), but they are aimed at more complex types of repetitiveness, such as versioned document collections, with a linear or tree versioning structure. Our case, without any versioning structure, is simpler to handle.
3.5 Local version
Not all the files handled by the applications will consist of genomes. Our filesystem can be configured to act on specific types of files, while treating the others in the usual way.
We use FUSE (https://github.com/libfuse/libfuse), a popular framework for the development of filesystems in user space, to build our prototype. Using the FUSE library we can add our own code to the processing of a number of system calls (stat, open, read, write, close, etc.). We then add specific methods that detect and treat differently the common files from the ones compressed by our method, redirecting to the usual system calls in one case, and to our compression or decompression methods in the other.
In this scheme, a process receives the system calls that involve files in a certain intervened path. Using the file’s extension and its particular path, the system decides whether to process it as a regular file (for example, by returning bytes from its contents in a read call), or to treat it in a special way (for example, by decompressing part of its content and returning it).
At the initial configuration time, the path that will be intervened by this file system (its mounting point) and the references to be used for compression are defined. The system can handle multiple references to compress different kinds of files, each associated with a particular subdirectory. It can also generate an artificial reference from the files in the directory, if none is given as a reference. When a file with the right extension is stored in one of those subdirectories, the system compresses it with the appropriate reference. When reading such a file, the decompressor is activated and the appropriate reference is loaded (if it was not already in RAM) in order to extract the desired portion of the file. The write calls on files with the right extension use, in a similar way, the appropriate compressor’s writing method.
The files compressed by this technique are then seen and manipulated by applications as if they were usual, uncompressed, files. Yet, the effective sizes on disk of the files stored in the filesystem are much smaller than the sizes seen by applications. This arrangement is shown in Figure 6.
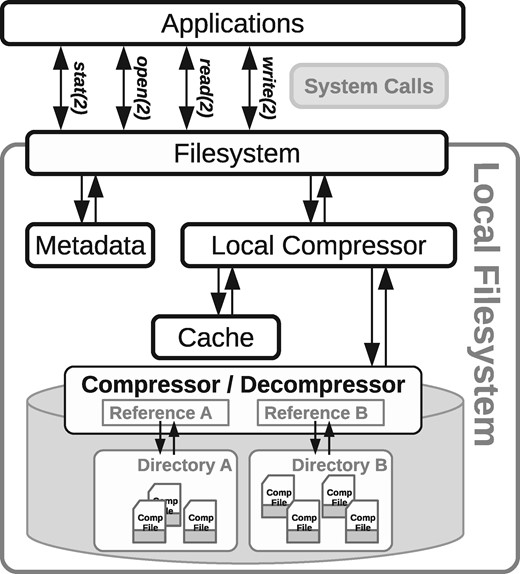
The local filesystem architecture
3.6 Client/server version
Using the local filesystem as a basis, we also build a version of the system based on the client/server model. In this mode, the client filesystem is installed and executed locally, but instead of storing the files on its own disk, it communicates with a remote server that stores the files in a centralized repository. Figure 7 displays the general architecture.
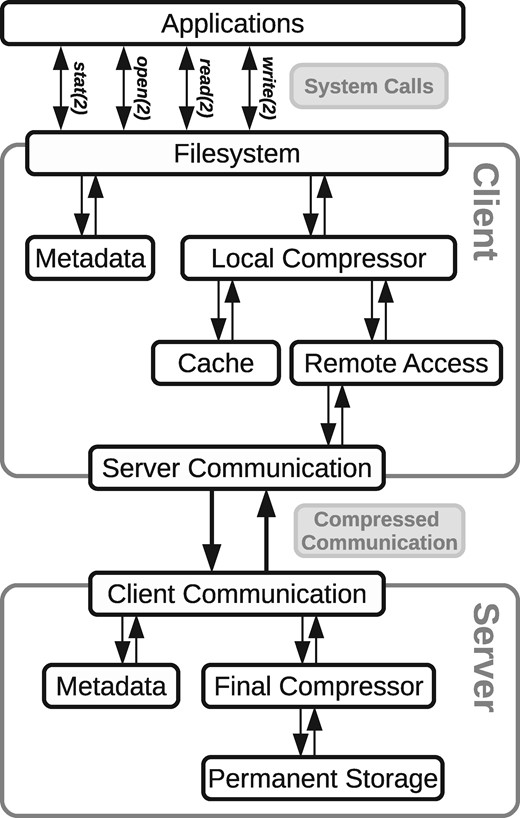
The client/server filesystem architecture
When a client communicates with the server for the first time, it sends its reference files, so that any further communication can proceed in compressed form. Server and client exchange whole files only.
The client intervenes the system calls as in the basic mode. When it needs to open a file, the file is transferred from the server in compressed form. Then the client stores the file in its own disk and handles it locally, as in the basic mode, until the file is closed. At this point, if the file was modified, it is sent back to the server in compressed form, to reduce network transfer time. The client also compresses the new files that are added to the filesystem, before sending them to the server.
Since the client may have little available memory for compression, it may use a sampled suffix array to (re)compress the file (recall Section 3). The server receives the files from the client, and if they were compressed with a sampled suffix array, it decompresses and recompresses them with the full suffix array, in order to obtain the best compression ratio. The next time the client requests the file, the optimally-compressed version will be sent. Note that both server and client can decompress a file compressed with the full or the sampled suffix array, as long as they share the same reference. Figure 8 shows the complete mechanism.
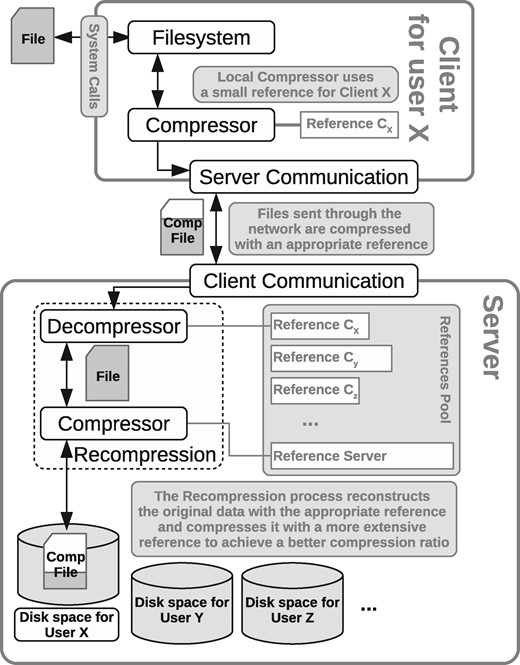
Complete client/server model of the platform
With minor changes, it is possible to optimize the writing process so that the client only sends the blocks that changed in the file, and the server only has to recompress those.
4 Results
We implemented our compressed file system using C++ and structures from the STL and the FUSE library, and compiled our codes with g++ 5.3.1 using the optimization flag -O3. All the experiments were done using a HP ProLiant DL380 G7 (589152-001) server, with two Quadcore Intel(R) Xeon(R) CPU E5620 @ 2.40GHz processors and a 96GB RAM, running version 2.6.34.9-69.fc13.x86 64 of Linux kernel. In the different experiments we evaluated the real elapsed time, and used that information to calculate the speed (in megabytes per second) of some of the processes.
4.1 Basic setup
In order to give two distant samples of the performance of whole-genome compression, we used two genome collections. The first is a set of 2504 fully assembled human genomes from Phase 3 of the 1000 Genomes Project (1000 Genomes Project Consortium et al., 2015) (http://www.internationalgenome.org), using the assembly HS37D5 as the reference. The second is a set of 1135 Arabidopsis thaliana genomes from the 1001 Genomes Project (https://1001genomes.org), using the TAIR10 assembly as the reference. We chose ten human and 200 A. thaliana genomes at random for the experiments. Human genomes have an approximate size of 3 GB, whereas the size of an A. thaliana genome is about 116 MB. Thus, a reference plus a full suffix array require about 15 GB for humans and 464 MB for A. thaliana.
If we just stored the genomes packing symbols in 2 bits per base [typical compressors do not reduce this space much further anyway (Biji and Achuthsankar, 2017)], the space required would be around 750 MB for human and 29 MB for A. thaliana genomes. Table 1 compares these sizes with the space we achieve with RLZ using the given reference genomes (with a full suffix array on the reference, for maximum compression) and averaging over the randomly chosen genomes. As it can be seen, our human compressed files are 85 times smaller than the original files, whereas for A. thaliana they are 37 times smaller. (While the compression of human genomes varies little from genome to genome, the A. thaliana dataset has more variance, and it could benefit from choosing more than one reference genome. For example, a set of 20 genomes chosen to best match the reference obtain a 58-fold compression.) Compression proceeds at 35–70 MB/s, whereas decompression proceeds at 100–390 MB/s (measuring MB of the original file). A whole human genome is then compressed in about 40 s, and recovered in about 30 s.
Mean sizes of the text, 2 bits per base and RLZ compressed files, and mean speed of compression and decompression (in MB/s) for the human and A. thaliana genomes used for the experiments
| Sequence | File size (1 byte/base) | Packed size (2 bits/base) | RLZ size | Compression speed | Decompression speed |
|---|---|---|---|---|---|
| Human | 3042 MB | 750 MB | 36 MB | 68.26 | 102.28 |
| A. thaliana | 116 MB | 29 MB | 3.1 MB | 36.59 | 385.38 |
| Sequence | File size (1 byte/base) | Packed size (2 bits/base) | RLZ size | Compression speed | Decompression speed |
|---|---|---|---|---|---|
| Human | 3042 MB | 750 MB | 36 MB | 68.26 | 102.28 |
| A. thaliana | 116 MB | 29 MB | 3.1 MB | 36.59 | 385.38 |
Mean sizes of the text, 2 bits per base and RLZ compressed files, and mean speed of compression and decompression (in MB/s) for the human and A. thaliana genomes used for the experiments
| Sequence | File size (1 byte/base) | Packed size (2 bits/base) | RLZ size | Compression speed | Decompression speed |
|---|---|---|---|---|---|
| Human | 3042 MB | 750 MB | 36 MB | 68.26 | 102.28 |
| A. thaliana | 116 MB | 29 MB | 3.1 MB | 36.59 | 385.38 |
| Sequence | File size (1 byte/base) | Packed size (2 bits/base) | RLZ size | Compression speed | Decompression speed |
|---|---|---|---|---|---|
| Human | 3042 MB | 750 MB | 36 MB | 68.26 | 102.28 |
| A. thaliana | 116 MB | 29 MB | 3.1 MB | 36.59 | 385.38 |
To compare with a plain compressor based on RLZ, we choose GDC2, the recent variant of Deorowicz et al. (2015). GDC2 compresses the human genomes of Table 1 to 11.2 MB and those of A. thaliana to 0.8 MB. This is roughly 3–4 times smaller than our compressed file. This factor is the price of pursuing a representation that can be directly accessed and modified: use of fixed-length numbers, partition the file into blocks, etc.
4.2 Tuning the block size
We tested several block sizes for the compressor. Figures 9 and 10 show the normalized compressed size, compression speed, and decompression speed, as a function of the block size, on the human genome HG00096 and A. thaliana genome CS78800. We tested with block sizes from 103 to 107 bytes, looking for a size that is sufficiently large to yield good space and time compression performance, and sufficiently small not to significantly compromise positional reads and writes on the files. The results are similar for all the human and A. thaliana genomes in our sample.
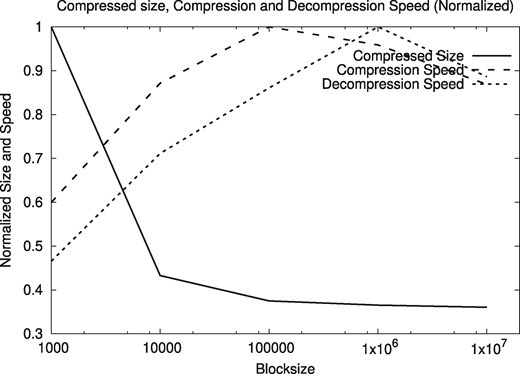
Normalized compressed size, compression and decompression speeds for varying block size, on the human genomes
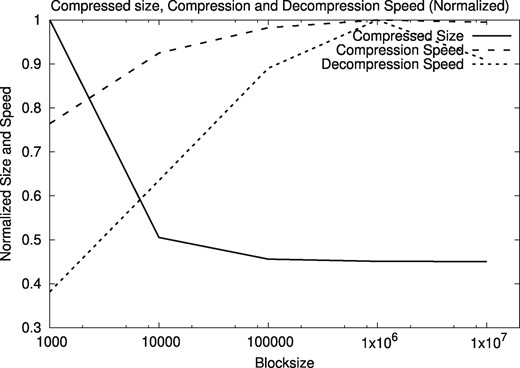
Normalized compressed size, compression and decompression speeds for varying block size, using A. thaliana sequences
Although the most usual operations in the filesystem are the addition of new text at the end of the file and the sequential decompression of the complete file, we also tested the effect of different block sizes in the times for random access in the compressed files. Our tests choose random positions from where segments of some fixed length are read. We averaged over 1000 starting positions, considering the extraction of text segments of length 100 KB, 1 MB and 10 MB for both human and A. thaliana genomes. The results are shown in Figures 11 and 12.
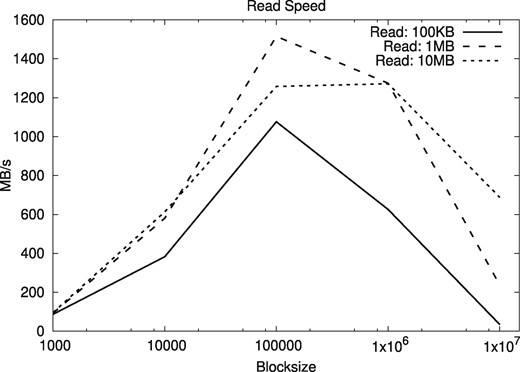
Random access speeds for segments of different lengths, as a function of the block size, using human genomes
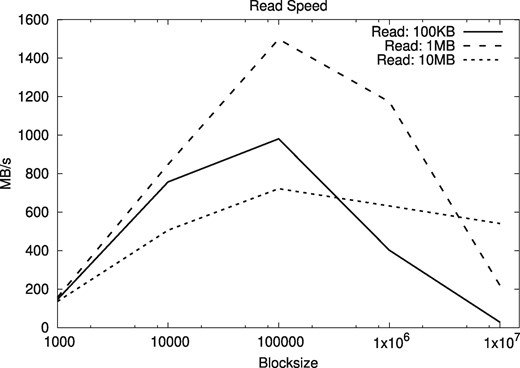
Random access speeds for segments of different lengths, as a function of the block size, using A. thaliana genomes
The results show that block sizes between 105 and 106 are adequate. We use blocks of size 105 (i.e. 100 KB) for the rest of the experiments. With this block size, both random and sequential access to the compressed files is roughly around 1 GB/s, and only 3–5 times slower than access to the plain files, whereas the space savings are of orders of magnitude.
4.3 Tuning the suffix array sampling
We use a reduced version of the suffix array to facilitate the usage of the client/server version. To reduce the size of the suffix array, we sample the text positions that are indexed. Smaller sampled suffix arrays worsen the compression ratios, however. Figures 13 and 14 show the resulting index sizes with different sampling frequencies, from 1 (i.e. the full suffix array) to 10 000 (i.e. storing one out of 10 000 text positions in the suffix array). The relative index size stabilizes at 0.2 because it counts the text.
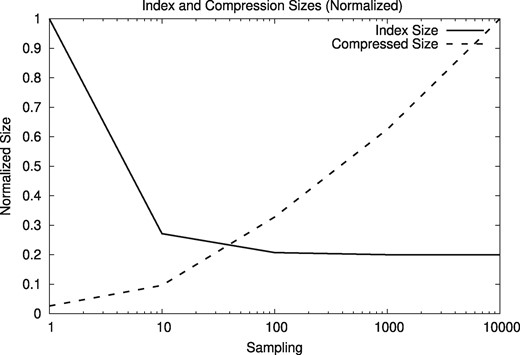
Normalized sizes of the index and the compressed files depending on the sampling frequency for human genomes
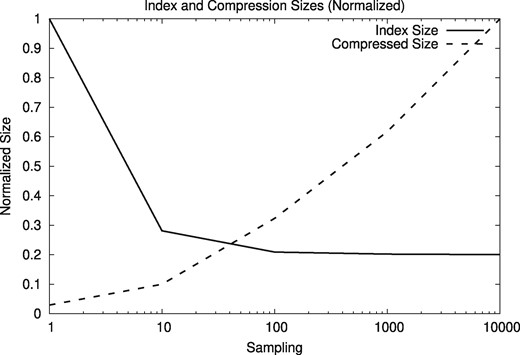
Normalized sizes of the index and the compressed files depending on the sampling frequency for A. thaliana genomes
Based on the results, we use a sampling frequency of 10 for the reference index. With that configuration, the size of the compressed human genome HG00096 increases from 36 MB to a still manageable 112 MB (27-fold compression), while reducing the effective index size from 15 GB to only 3.8 GB, enough to run smoothly on a desktop PC. Using the A. thaliana genome CS78800 and a sampling frequency of 10 increases the size of the compressed sequence from 2 MB to around 6.4 MB (still 18-fold compression), while reducing the index size from 464 MB to 162 MB.
We remind that, in a client/server scenario, those weaker compression ratios will be obtained only when the client compresses the files temporarily with a reduced suffix array. Those will be recompressed with a full suffix array when they arrive to the server, and used in this better-compressed form when the client requests the files again.
4.4 Remote access to compressed files
To test the efficiency of the remote access of compressed files compared to the usage of decompressed files, we measure the time to compress, transfer the compressed files, and decompress those files locally. Table 2 compares those times with the cost of transferring the uncompressed text, for different prefixes of the human genome HG00096. It can be seen that, when considering files over hundreds of MBs, remotely accessing the compressed data is about 4–5 times faster accessing the corresponding uncompressed data. Moreover, these times take into account the compression process prior to the transfer, but in most cases a file is compressed only once and accessed many times. If we consider only the transfer times of compressed files and the local decompression, the gain due to using compression is even more significant, 8–12 times faster.
Times in milliseconds for compression, transfer, decompression, and total for RLZ compressed files, transfer time for uncompressed files using a typical 100 Mb/s speed network, and the time for locally copying the text files
| Filesize (MB) | RLZ | RLZ | RLZ | RLZ | Uncompressed (100 Mb/s network) | Copy time |
|---|---|---|---|---|---|---|
| Compression | Transfer | Decompression | Total Time | |||
| 3 | 91 | 663 | 7 | 761 | 842 | 5 |
| 30 | 603 | 1144 | 59 | 1806 | 3523 | 35 |
| 300 | 5315 | 2201 | 795 | 8311 | 34 921 | 597 |
| 3000 | 42 411 | 4569 | 28 676 | 75 656 | 271 604 | 24 018 |
| Filesize (MB) | RLZ | RLZ | RLZ | RLZ | Uncompressed (100 Mb/s network) | Copy time |
|---|---|---|---|---|---|---|
| Compression | Transfer | Decompression | Total Time | |||
| 3 | 91 | 663 | 7 | 761 | 842 | 5 |
| 30 | 603 | 1144 | 59 | 1806 | 3523 | 35 |
| 300 | 5315 | 2201 | 795 | 8311 | 34 921 | 597 |
| 3000 | 42 411 | 4569 | 28 676 | 75 656 | 271 604 | 24 018 |
Times in milliseconds for compression, transfer, decompression, and total for RLZ compressed files, transfer time for uncompressed files using a typical 100 Mb/s speed network, and the time for locally copying the text files
| Filesize (MB) | RLZ | RLZ | RLZ | RLZ | Uncompressed (100 Mb/s network) | Copy time |
|---|---|---|---|---|---|---|
| Compression | Transfer | Decompression | Total Time | |||
| 3 | 91 | 663 | 7 | 761 | 842 | 5 |
| 30 | 603 | 1144 | 59 | 1806 | 3523 | 35 |
| 300 | 5315 | 2201 | 795 | 8311 | 34 921 | 597 |
| 3000 | 42 411 | 4569 | 28 676 | 75 656 | 271 604 | 24 018 |
| Filesize (MB) | RLZ | RLZ | RLZ | RLZ | Uncompressed (100 Mb/s network) | Copy time |
|---|---|---|---|---|---|---|
| Compression | Transfer | Decompression | Total Time | |||
| 3 | 91 | 663 | 7 | 761 | 842 | 5 |
| 30 | 603 | 1144 | 59 | 1806 | 3523 | 35 |
| 300 | 5315 | 2201 | 795 | 8311 | 34 921 | 597 |
| 3000 | 42 411 | 4569 | 28 676 | 75 656 | 271 604 | 24 018 |
In our experiments we used a 100 Mb/s network (which, in practice, allows the transfer of large files at about 12 Mb/s). A network with higher transfer speeds would benefit more the remote access of uncompressed files. However, the time to transfer the files will always be greater than the time needed to make a local copy of those files, so we also include the cost of that operation in Table 2. In the case of compressed files, the decompression time (which includes the local creation of the decompressed text file) could be considered a similar minimum time. We can see that, in fact, the decompression times are only slightly higher than those of making a simple copy of the data. Therefore, even using a significantly faster network, we can access the data at comparable times and preserve the benefits of reduced file sizes using this kind of compression.
4.5 Sequence reads
To test the case of storing a collection of non-assembled reads, we use a set of human embryo development RNA-seq reads from the NIH Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra), the dataset SRR445718. This set contains around 3.3 Gbp and the full FASTA file is about 5.4 GB, including metadata. We use our reference generation method of Section 3.4, producing a reference of 5% of the size of the sequence data (i.e. 160 MB) formed by segments of length 5000. Using this reference and the metadata compression system with LZMA, we reduced the sequences to 14.1% of its original size. In this FASTA file, approximately 40% of the text consists of metadata. This is significantly higher than in the previous test cases, in which practically all the text consists of genetic sequences. Without considering the metadata, the space of the sequence data is reduced to 11.5%.
The compressed file can be accessed randomly and sequentially at a speed comparable to our fully-assembled genomes, around 800 MB/s.
5 Discussion
We have described a compressed filesystem for genome collections, which exploits Relative Lempel-Ziv compression to obtain reductions in file sizes of 1–2 orders of magnitude, while allowing applications to transparently access and modify the files through the usual filesystem calls. In addition, a client/server architecture frees the clients from storing the files themselves, which are stored in a centralized server that provides stronger compression and transfers the files to and from the clients in compressed form, thereby reducing network transfer times by an order of magnitude as well.
The most important line of future work, to let the system scale up, is to automate the choice of the references. In the current system, these are defined manually by the user, one per directory where the genome files are stored. Alternatively, the system can build an artificial reference from an initial set of given files, but this process is also launched manually. Instead, the filesystem could choose and update the references on the fly as the sequence data evolves.
There has been some work on how to choose the references (Deorowicz and Grabowski, 2011; Gagie et al., 2016; Kuruppu et al., 2011; Liao et al., 2016), showing that even randomly chosen chunks from the files work well. In our case, a simple scheme like choosing the first stored file as a reference, and appending chunks of incoming files that do not compress well with respect to the current reference, is likely to produce a suitable reference without user intervention. Further, a central server storing genomes from multiple clients could use the same references for many clients, thereby further reducing the storage costs.
A second line of work is to further reduce storage needs for the references and suffix arrays. First, we could use alphabet mapping so as to store the references using exactly bits per symbol (where σ is the alphabet size) instead of always 8 (i.e. bytes). On DNA sequences, this would use 2 bits per base and thus reduce by 4 the space to store the reference. Similarly, the full suffix array could be replaced by an FM-index (Ferragina and Manzini, 2005), which would require about 2 bits per entry (instead of 32). This structure has already been shown to offer competitive performance in practical applications (see, e.g. http://bowtie-bio.sourceforge.net/bowtie2).
Finally, we may integrate some features of more recent variants of RLZ [e.g. by Deorowicz et al. (2015)] with our filesystem, aiming to obtain higher compression speeds or better compression ratios while retaining direct access and the possibility to modify the compressed file.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}