





Abstract
The progress of High Throughput Sequencing (HTS) technologies and the reduction in the sequencing costs are such that Whole Genome Sequencing (WGS) could replace many traditional laboratory assays and procedures. Exploiting the volume of data produced by HTS platforms requires substantial computing skills and this is the main bottleneck in the implementation of WGS as a routine laboratory technique. The way in which the vast amount of results are presented to researchers and clinicians with no specialist knowledge of genome sequencing is also a significant issue.
Here we present TORMES, a user-friendly pipeline for WGS analysis of bacteria from any origin generated by HTS on Illumina platforms. TORMES is designed for non-bioinformatician users, and automates the steps required for WGS analysis directly from the raw sequence data: sequence quality filtering, de novo assembly, draft genome ordering against a reference, genome annotation, multi-locus sequence typing (MLST), searching for antibiotic resistance and virulence genes, and pangenome comparisons. Once the analysis is finished, TORMES generates and interactive web-like report that can be opened in any web browser and shared and revised by researchers in a simple manner. TORMES can be run by using very simple commands and represent a quick an easy way to perform WGS analysis.
TORMES is free available at https://github.com/nmquijada/tormes.
Supplementary data are available at Bioinformatics online.
1 Introduction
The development of High-Throughput DNA Sequencing (HTS) technologies and the fall in the cost of sequencing have irreversibly changed how bacteria are investigated (Carriço et al., 2018). High-throughput bacterial genome sequencing (also called ‘whole genome sequencing’, WGS) is one of the most popular HTS applications and may replace various traditional molecular and laboratory tests (Taboada et al., 2017). The ability to generate bacterial draft genome sequences routinely has diverse applications, including the analysis of clinical and laboratory strains and mutants, pathogen surveillance, outbreak detection and investigations of antibiotic resistance (ABR) (Harris et al., 2013; Holt et al., 2012; Howden et al., 2011; Taboada et al., 2017).
The most recent bench-top HTS platforms allow bacterial draft genome sequences to be obtained in the laboratory within a few hours or days (Deurenber et al., 2017). Illumina’s HTS platform has become one of the most popular: they generate millions of reads ∼100 to ∼300 bp long with low error rates (<1%) such that bacterial draft genomes can be acquired with high accuracy and coverage (Deng et al., 2016; Goodwin et al., 2016).
WGS is faster and cheaper than traditional methods (such as pulsed-field gel electrophoresis, Sanger sequencing of housekeeping genes for MLST or phenotypic testing) and offers more information: the entire genomic content of the target bacterium (Köser et al., 2014; Ronholm et al., 2016; Taboada et al., 2017). This approach is used by several organizations, and in particular the Food and Drug Administration (FDA) and Center for Disease Control and Prevention (CDC), but its application as a routine laboratory practice is still limited (Carriço et al., 2018; Rantsiou et al., 2018). Although the hardware required for WGS are is now affordable, the analysis of the huge amount of data produced by HTS platforms requires advanced computational skills not available to every microbiology laboratory; this is one of the main impediments to the application of WGS (Logares et al., 2012; Sekse et al., 2017). Diverse specialized software for WGS analysis are being developed and adapted in this technological environment, and indeed bacterial bioinformatics is a constantly developing field and a challenge for researchers without a bioinformatics background (Carriço et al., 2018). The routine application of WGS requires cheap, user-friendly techniques that can be used on-site by personnel not specialized in big data management (Hyeon et al., 2018).
Here we present TORMES, an open-source, user-friendly, command-line pipeline for conducting WGS analysis of HTS data produced by Illumina platforms from a set of bacteria. TORMES was designed for non-bioinformatician scientists: automates the steps of the bioinformatic analysis, including sequence quality filtering, de novo assembly, draft genome ordering against a reference, genome annotation, MLST, searches for antibiotic resistance and virulence genes and pangenome comparisons. This can be done directly from the raw sequencing data without the need for an internet connection, by following very simple instructions.
Condensing large amounts of data in a format that can be understood by researchers and clinicians with no specialist knowledge of genome sequencing has been identified as a major issue for bacterial genomics (Köser et al., 2012). TORMES stores every file generated during the process and once the analysis is done, the results are summarized in an interactive web-like report that can be revised, shared and compared in an ergonomic manner.
The report is generated in R environment by using an automatically generated RMarkdown code file, unique for each analysis. It is also kept in a separate folder to allow more specialist users to deepen the analysis and modify the code for user-specific reports.
TORMES can be used with an unlimited number of samples and was tested on hundreds of bacterial genomes in isolates of numerous species (including Escherichia, Salmonella, Clostridium and Klebsiella spp.) from different origins (clinical, fecal, animal and food-related an environmental) sequenced on Illumina platforms (as an approximation, TORMES analysis with default options of 100 ∼3.5 Mbp length genomes with ∼50× sequencing depth lasted 16 h on a 124 GB RAM 32 cores computer).
2 Materials and methods
2.1 Overview of TORMES pipeline
TORMES (named after the river that flows through the city of Salamanca, Spain, in honor of the eight hundredth anniversary of the city’s University) is a pipeline for bacterial WGS analysis directly from raw paired-end sequences obtained from Illumina HTS platforms. The TORMES pipeline includes several steps conducted by different software and that are stored in separate directories for further user usage (summarized in Fig. 1). First, possible remaining sequencing adaptors are removed from raw sequencing reads by using Trimmomatic (Bolger et al., 2014) that are further quality filtered by using Prinseq (Schmieder and Edwards, 2011), Sickle (Joshi and Fass, 2011) or Trimmomatic. The chose of the software to perform the quality filtering, the minimum mean quality score or the minimum length of the raw reads to overcome the filtering can be made by using the --filtering, –q/--quality and --min_len options. Kraken (Wood and Salzberg, 2014) is used to classify the reads taxonomically as an additional quality control. The reads that pass the quality controls are then assembled de novo into a draft genome. Genome assembly can be carried out with SPAdes (Bankevich et al., 2012) or Megahit (Li et al., 2015) by using the --assembler option. TORMES is designed to work with any bacterial genome; the de novo assembly approach is the method of choice for any new bacterium or new strain of a well-known bacterium (Loman et al., 2012). The quality of the assemblies is evaluated by QUAST (Gurevich et al., 2013) and contigs below 200 bp long are discarded. If a closely related genome is available, the contigs in the draft genome can be ordered against it with the option –r/--reference using progressiveMauve (Darling et al., 2010). Draft genomes (ordered or not) are then annotated using Prokka (Seemann, 2014). Prokka generates several text-format files per genome annotated that TORMES stores in separate directories for each sample (all included in the main ‘annotation’ directory, see below). Functional assignation of the predicted genes relies in the information stored in the annotation databases. Standard Prokka installation comes with small test databases that may be insufficient for genome analysis. Users are encouraged to increase and customize the annotation databases by following the instructions stated at the Prokka repository (https://github.com/tseemann/prokka). The ‘gff’ files generated with Prokka are used for a pangenome comparison between the samples using Roary and based on the presence/absence of predicted genes (Page et al., 2015). Pangenome distance trees and summary figures are generated by FastTree (Price et al., 2009) and roary2svg (T. Seemann, https://github.com/sanger-pathogens/Roary/blob/master/contrib/roary2svg/roary2svg.pl), respectively. Pangenome analysis can be skipped by using the --no_pangenome option. MLST is performed with the mlst software (T. Seemann, https://github.com/tseemann/mlst) and the PubMLST database (Jolley and Maiden, 2010), although the analysis can be disabled by using the --no_mlst option. The draft genome is screened for antibiotic resistance (ABR) and virulence genes using BLASTn (Zhang et al., 2000) and ABRicate (T. Seemann, https://github.com/tseemann/abricate) against ResFinder (Zankari et al., 2012), CARD (McArthur et al., 2013) and ARG-ANNOT (Gupta et al., 2014) or the Virulence Factors Database (VFDB) (Chen et al., 2004), respectively. Instructions for the development of custom-specific gene databases to be included in the TORMES pipeline can be addressed in TORMES repository. Several steps, notably the pangenome analysis and the genome ordering, are time and memory consuming. If the user is not interested in this data, these analyses can be avoided by enabling the --fast option, that also uses MegaHit for genome assembly.
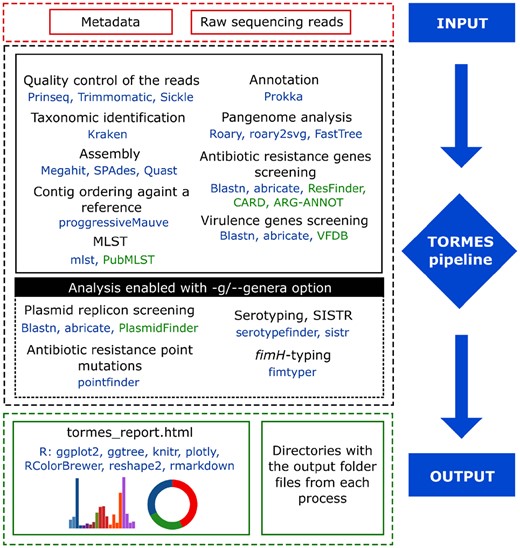
Summary of the TORMES analysis pipeline. Software and R packages appear in blue and databases in green
TORMES will work with any set of bacteria from any species and origin. More extensive analyses for Escherichia and Salmonella can be enabled by using the -g/--genera option (followed by ‘Escherichia’ or ‘Salmonella’). This allows identification of plasmid replicons using the PlasmidFinder database (Carattoli et al., 2014), detection of point mutations that can cause ABR using PointFinder (Zankari et al., 2017) and serotyping analysis using SerotypeFinder (Joensen et al., 2015) for Eschericha or SISTR (Yoshida et al., 2016) for Salmonella. FimH-typing for Eschericha is also possible by using FimTyper (Roer et al., 2017).
All the files generated during the WGS analysis are stored in the directory specified by the option -o/--output. The output directory harbors different directories regarding the processes (Fig. 1). The RMarkdown code file (‘tormes_report.Rmd’), used for the creation of the summary report, is also stored, so users can freely reproduce, modify and generate a user-specific report adjusted to their requirements (instructions can be found in the TORMES repository). The report is generated in R environment (R Development Core Team, 2008) using R packages: ggplot2 (Wickham, 2009), ggtree (Yu et al., 2017), knitr (Xie, 2015), plotly (Sievert et al 2017), RColorBrewer (Neuwirth and Brewer, 2014), reshape2 (Wickham, 2007) and rmarkdown (Allaire, 2015). The web-like report summarizes the results of the TORMES analysis (an example is provided in https://nmquijada.github.io/tormes/files/). The file is small (a few MB) allowing easy exchange of information between groups; the report can be opened in any web browser and all tables and figures contained can be copied or downloaded.
The software items listed above are the backbone of TORMES and users are encouraged to cite them (and their version) together with the present article when using TORMES. A summary of the software used by TORMES is included in Supplementary File 1.
2.2 Equipment and software setup
TORMES pipeline was built using GNU bash v.4.2.46 (http://www.gnu.org/software/bash/), and R v.3.4.3 and can be run on any UNIX system computer. The TORMES pipeline integrates the GNU parallel (Tange, 2011) allowing efficient use of the computer (using the -t/--threads option). HTS data require storage space: as an approximation, the TORMES output will double the size of the input data. In the analysis reported here, 1.9 GB of input data (gzipped fastq files) generated a results directory of 3.9 GB.
The TORMES pipeline is freely available in https://github.com/nmquijada/tormes, with a manual for its use. TORMES pipeline and all the required software and dependencies can be automatically installed by using conda.
2.3 Benefits of open-source project
TORMES software, the instructions for it use and the report generated in the case study can be found in https://github.com/nmquijada/tormes. Bacterial bioinformatics is developing rapidly and the availability of open code and tools is crucial for the scientific community to benefit from these developments. New software and tools emerge almost daily and our intention is for the TORMES project to be updated efficiently. The –g/--genera option was included to allow TORMES to perform extra analyses of particular bacteria (‘Escherichia’ and ‘Salmonella’ options are currently available). TORMES is intended to be a networking project with users providing their feedback and personal experience so that TORMES can become a more complete pipeline including as many analyses and genera as possible.
3 Results
3.1 Case study: analysis of ten Salmonalla spp. by using TORMES
TORMES has been tested on hundreds of bacterial genomes from different species and from different sources. As a case study, we report the analysis of ten Salmonella spp. that were isolated from food illegally imported into the EU. DNA was extracted and sequenced on an Illumina MiSeq platform (Supplementary File 2) and the raw fastq files were directly submitted to TORMES, with no other bioinformatic treatment, as follows:
tormes --metadata salmonella_metadata.txt
--output Salmonella_TORMES_2018 --reference
S_enterica-CT02021853.fasta --threads 32 --genera Salmonella
The file parsed to the --metadata option included information regarding each isolate (name, location of the reads in the computer and extra metadata). The analysis lasted 1 h in a 124 GB RAM, 32 cores computer and around 3 h in a 16 GB RAM 4 cores laptop. TORMES summarized the results in an interactive web-like file that can be visualized in https://nmquijada.github.io/tormes/files/. Raw sequencing data is stored in TORMES repository for the users to download, reproduce and compare the analysis.
Quality control generated 542 482 to 888 038 reads per sample (86% of reads survived overall), that were assembled yielding 4.67 ± 0.06 Mbp-long draft genomes formed by 41 to 188 contigs, with mean N50 of 128 ± 78 kbp and mean sequencing depth of 37 ± 5× (Supplementary File 3). MLST and serotyping revealed that all isolates belonged to subspecies enterica and to six different ST and serovars: three ST279 serovar I 4,[5], 12: d:-, two ST4 serovar Montevideo, two ST64 serovar Anatum, one ST11 serovar enteritidis, one ST23 serovar Oranienburg and one ST45 serovar Newport (Supplementary File 3). Pangenome analysis showed 5811 different genes overall, where 3602 of the genes (62% of the total of genes found) were common to all the samples (Supplementary File 4).
The isolates were screened for ABR genes (Fig. 2) with ResFinder, CARD and ARG-ANNOT databases. Isolates MS0498 and MS0499 (both ST279 serovar Montevideo) harbored qnrB, associated with resistance to quinolones. MS0496, MS0498 and MS0499 (all ST279) harbored the aminoglycoside resistance gene AAC(6)-Iaa, and all the other isolates harbored AAC(6)-Iy. All the isolates harbored the genes related to the multidrug and metal efflux pump complex MdsABC (golS, mdsA, mdsB and mdsC). Mutation T57S in ParC conferring resistance to nalidixic acid and ciprofloxacin was detected in all isolates but MS0500 (ST11). No plasmids nor single nucleotide mutations known for causing resistance in gyrA, gyrB, parE, pmrA or pmrB were found. Eighty-six to 96 virulence genes from the Virulence Factor Database were found per isolate (Supplementary File 5). Most of the virulence genes, including type-III secretion system genes, were found in all the isolates. MS0500 (ST11) was the only sample carrying sodCl and lpfD genes, related to the stress response and adherence, respectively. MS0500 (ST11), MS0495 and MS0497 (both ST4) harbored ratB which is involved in intestinal colonization and persistence. The cytolethal distending toxin gene cdtB was found in MS0495, MS0497 and MS0501 (ST23). The ST64 samples MS0502 and MS0503 harbored tcpC, a gene important in immune evasion and that promotes renal tissue damage.
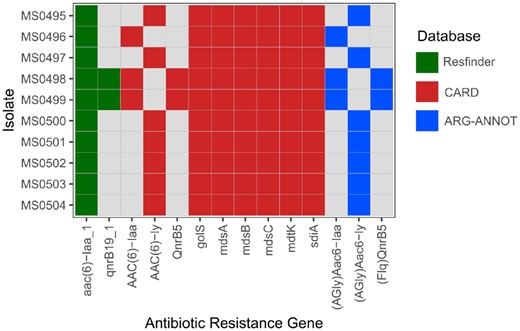
Presence of antibiotic resistance genes in the samples included in the case study determined using different databases: Resfinder (green), CARD (red) and ARG-ANNOT (blue)
Salmonella is an enteric pathogen that causes a range of diseases in humans with high associated morbidity and mortality, (Bula-Rudas et al., 2015; Nuccio and Bäumler, 2014). Multidrug resistant Salmonella spp. are a public health concern and its rapid identification and investigation is critical making its quick identification and investigation is widely important (Mourão et al., 2014; Ziech et al., 2016). The study shows the potential of TORMES to perform quick and in-depth WGS analysis directly from raw sequencing data by using simple commands.
4 Discussion
HTS has transformed microbiology (De Filippis et al., 2018). Rapid and low-cost genome sequencing is such that WGS may replace many current laboratory tests and methods (Ronholm et al., 2016). Scientists in academia, the regulatory sector and industry increasingly recognize WGS as the method of choice for basic research and epidemiological investigations (Rantsiou et al., 2018). However, the use of WGS as a routine laboratory technique requires overcoming existing challenges and limitations. Bioinformatic analysis is the main bottleneck in WGS studies, and is required due to handle the large amounts of data generated by HTS platforms (Oniciuc et al., 2018). Presenting and sharing the substantial volumes of information with technicians and clinicians with no specialist knowledge of genome sequencing can be problematic (Logares et al., 2012). Standardized and user-friendly software and pipelines make WGS analysis more accessible, even to those without bioinformatic training (De Filippis et al., 2018).
We developed TORMES, a bioinformatic pipeline for direct WGS analysis of raw sequencing data from Illumina HTS platforms. TORMES code is open, and with the software and databases versions utilized by the users, provide third parties with all they need to track the progress of the analysis. If the data and the underlying code used for the analysis are not made available, the ability of others to reproduce and build upon the analysis will obviously be limited (Schloss, 2018). ‘Best practice’ recommendations for making of research software robust consider the availability of code essential for experiment reproducibility and replicability (Taschuk and Wilson, 2017).
Several web tools, such as RAST (Aziz et al., 2008), PATRIC (Wattam et al., 2014) and MicroScope (Vallenet et al., 2013) have been developed in the last years, and integrate user-friendly interfaces that represent very powerful tools for the analysis and comparison of WGS data. However, analyses are preformatted and do not allow user customization and they require the data to be uploaded into their servers. TORMES is designed to run in your own computer without the need of internet connection, access accounts or extra requirements. TORMES integrates Prokka, which is the gold command-line application for bacterial genome annotation. Prokka starts from an assembled genome and it allows annotation databases customization that may lead to the inclusion of rare taxa that usually lacks on external databases (Rachid et al., 2013). TORMES expands the spectrum of Prokka as it starts directly from raw sequencing data and include additional analysis, such as contig ordering against a reference, pangenome analysis based on the presence/absence of accessory genes and core genome distances, detection of point mutations and subtyping analysis (such as MLST, serotyping and fimH-typing). TORMES can be run using very simple commands and be the method of choice for researches lacking strong bioinformatic background to perform WGS analysis of a set of bacteria.
Advanced genomics and bioinformatics systems have proven their worth in reducing response times to emerging disease outbreaks. They provide substantial socioeconomic benefits in terms of improved public health, reduced health care costs and avoidance of loss of productivity due to illness (Scharff et al., 2016). In silico serotype prediction methods, such as SISTR (Yoshida et al., 2016) and SerotypeFinder (Joensen et al., 2015), both implemented in TORMES for S. enterica and E. coli, respectively, are substantially cheaper and easier than conventional antibody-based serotype determinations (Taboada et al., 2017). In addition, TORMES allows the quick detection of antibiotic resistance (including point mutations in S. enterica and E. coli) and virulence genes, through exploiting curated and regularly updated databases, such as ResFinder, CARD, ARG-ANNOT and VFDB. Strengthening ABR surveillance worldwide is critical, and required for informing global strategies, monitoring the effectiveness of public health interventions and detecting new trends and emerging threats (Oniciuc et al., 2018). Finally, TORMES facilitates the interpretation and exchange of information, as results are automatically summarized in an interactive web-like report.
5 Conclusion
The implementation of WGS has enormous potential for bacterial research and its development is critically linked to the public availability of both genomic data and analysis tools. Regularly updated open-source and user-friendly pipelines and software, such as TORMES, may help to unleash the potential of bacterial bioinformatics and make WGS a feasible tool for the research community.
Acknowledgements
NMQ received a PhD fellowship from the Spanish National Institute for Agriculture and Food Research and Technology (INIA, Ministerio de Economía, Industria y Competitividad; fellowship FPI2014-020).
Funding
This study was co-funded by The Spanish Ministry of Economy, Industry and Competitiveness (MINECO; AGL2016-74882-C3), the Junta de Castilla y León (JCyL; GRS 1780/A/18) and the European Regional Development Fund (ERDF).
Conflict of Interest: none declared.
References
R Development Core Team. (


{kind=link}
{kind=link}