





Abstract
The ability to generate massive amounts of sequencing data continues to overwhelm the processing capability of existing algorithms and compute infrastructures. In this work, we explore the use of hardware/software co-design and hardware acceleration to significantly reduce the execution time of short sequence alignment, a crucial step in analyzing sequenced genomes. We introduce Shouji, a highly parallel and accurate pre-alignment filter that remarkably reduces the need for computationally-costly dynamic programming algorithms. The first key idea of our proposed pre-alignment filter is to provide high filtering accuracy by correctly detecting all common subsequences shared between two given sequences. The second key idea is to design a hardware accelerator that adopts modern field-programmable gate array (FPGA) architectures to further boost the performance of our algorithm.
Shouji significantly improves the accuracy of pre-alignment filtering by up to two orders of magnitude compared to the state-of-the-art pre-alignment filters, GateKeeper and SHD. Our FPGA-based accelerator is up to three orders of magnitude faster than the equivalent CPU implementation of Shouji. Using a single FPGA chip, we benchmark the benefits of integrating Shouji with five state-of-the-art sequence aligners, designed for different computing platforms. The addition of Shouji as a pre-alignment step reduces the execution time of the five state-of-the-art sequence aligners by up to 18.8×. Shouji can be adapted for any bioinformatics pipeline that performs sequence alignment for verification. Unlike most existing methods that aim to accelerate sequence alignment, Shouji does not sacrifice any of the aligner capabilities, as it does not modify or replace the alignment step.
Supplementary data are available at Bioinformatics online.
1 Introduction
One of the most fundamental computational steps in most bioinformatics analyses is the detection of the differences/similarities between two genomic sequences. Edit distance and pairwise alignment are two approaches to achieve this step, formulated as approximate string matching (Navarro, 2001). Edit distance approach is a measure of how much two sequences differ. It calculates the minimum number of edits needed to convert a sequence into the other. The higher the edit distance the more different the sequences from one another. Commonly allowed edit operations include deletion, insertion and substitution of characters in one or both sequences. Pairwise alignment is a measure of how much the sequences are alike. It calculates the alignment that is an ordered list of characters representing possible edit operations and matches required to change one of the two given sequences into the other. As any two sequences can have several different arrangements of the edit operations and matches (and hence different alignments), the alignment algorithm usually involves a backtracking step. This step finds the alignment that has the highest alignment score (called optimal alignment). The alignment score is the sum of the scores of all edits and matches along the alignment implied by a user-defined scoring function. The edit distance and pairwise alignment approaches are non-additive measures (Calude et al., 2002). This means that if we divide the sequence pair into two consecutive subsequence pairs, the edit distance of the entire sequence pair is not necessarily equivalent to the sum of the edit distances of the shorter pairs. Instead, we need to examine all possible prefixes of the two input sequences and keep track of the pairs of prefixes that provide an optimal solution. Enumerating all possible prefixes is necessary for tolerating edits that result from both sequencing errors (Fox et al., 2014) and genetic variations (McKernan et al., 2009). Therefore, the edit distance and pairwise alignment approaches are typically implemented as dynamic programming algorithms to avoid re-examining the same prefixes many times. These implementations, such as Levenshtein distance (Levenshtein, 1966), Smith–Waterman (Smith and Waterman, 1981) and Needleman–Wunsch (Needleman and Wunsch, 1970), are inefficient as they have quadratic time and space complexity [i.e. O(m2) for a sequence length of m]. Many attempts were made to boost the performance of existing sequence aligners. Despite more than three decades of attempts, the fastest known edit distance algorithm (Masek and Paterson, 1980) has a running time of O(m2/log2m) for sequences of length m, which is still nearly quadratic (Backurs and Indyk, 2017). Therefore, more recent works tend to follow one of two key new directions to boost the performance of sequence alignment and edit distance implementations: (i) accelerating the dynamic programming algorithms using hardware accelerators. (ii) Developing filtering heuristics that reduce the need for the dynamic programming algorithms, given an edit distance threshold.
Hardware accelerators are becoming increasingly popular for speeding up the computationally expensive alignment and edit distance algorithms (Al Kawam et al., 2017; Aluru and Jammula, 2014; Ng et al., 2017; Sandes et al., 2016). Hardware accelerators include multi-core and single instruction multiple data (SIMD) capable central processing units (CPUs), graphics processing units (GPUs) and field-programmable gate arrays (FPGAs). The classical dynamic programming algorithms are typically accelerated by computing only the necessary regions (i.e. diagonal vectors) of the dynamic programming matrix rather than the entire matrix, as proposed in Ukkonen’s banded algorithm (Ukkonen, 1985). The number of the diagonal bands required for computing the dynamic programming matrix is 2E + 1, where E is a user-defined edit distance threshold. The banded algorithm is still beneficial even with its recent sequential implementations as in Edlib (Šošić and Šikić, 2017). The Edlib algorithm is implemented in C for standard CPUs and it calculates the banded Levenshtein distance. Parasail (Daily, 2016) exploits both Ukkonen’s banded algorithm and SIMD-capable CPUs to compute a banded alignment for a sequence pair with a user-defined scoring function. SIMD instructions offer significant parallelism to the matrix computation by executing the same vector operation on multiple operands at once. The multi-core architecture of CPUs and GPUs provides the ability to compute alignments of many sequence pairs independently and concurrently (Georganas et al., 2015; Liu and Schmidt, 2015). GSWABE (Liu and Schmidt, 2015) exploits GPUs (Tesla K40) for highly parallel computation of global alignment with a user-defined scoring function. CUDASW++ 3.0 (Liu et al., 2013) exploits the SIMD capability of both CPUs and GPUs (GTX690) to accelerate the computation of the Smith–Waterman algorithm with a user-defined scoring function. CUDASW++ 3.0 provides only the optimal score, not the optimal alignment (i.e. no backtracking step). Other designs, for instance FPGASW (Fei et al., 2018), exploit the very large number of hardware execution units in FPGAs (Xilinx VC707) to form a linear systolic array (Kung, 1982). Each execution unit in the systolic array is responsible for computing the value of a single entry of the dynamic programming matrix. The systolic array computes a single vector of the matrix at a time. The data dependencies between the entries restrict the systolic array to computing the vectors sequentially (e.g. top-to-bottom, left-to-right or in an anti-diagonal manner). FPGA accelerators seem to yield the highest performance gain compared to the other hardware accelerators (Banerjee et al., 2018; Chen et al., 2016; Fei et al., 2018; Waidyasooriya and Hariyama, 2015). However, many of these efforts either simplify the scoring function, or only take into account accelerating the computation of the dynamic programming matrix without providing the optimal alignment as in Chen et al. (2014), Liu et al. (2013) and Nishimura et al. (2017). Different and more sophisticated scoring functions are typically needed to better quantify the similarity between two sequences (Henikoff and Henikoff, 1992; Wang et al., 2011). The backtracking step required for the optimal alignment computation involves unpredictable and irregular memory access patterns, which poses a difficult challenge for efficient hardware implementation.
Pre-alignment filtering heuristics aim to quickly eliminate some of the dissimilar sequences before using the computationally expensive optimal alignment algorithms. There are a few existing filtering techniques, such as the Adjacency Filter (Xin et al., 2013), which is implemented for standard CPUs as part of FastHASH (Xin et al., 2013). SHD (Xin et al., 2015) is a SIMD-friendly bit-vector filter that provides higher filtering accuracy compared to the Adjacency Filter. GRIM-Filter (Kim et al., 2018) exploits the high memory bandwidth and the logic layer of 3D-stacked memory to perform highly-parallel filtering in the DRAM chip itself. GateKeeper (Alser et al., 2017a) is designed to utilize the large amounts of parallelism offered by FPGA architectures. MAGNET (Alser et al., 2017b) shows a low number of falsely accepted sequence pairs but its current implementation is much slower than that of SHD or GateKeeper. GateKeeper (Alser et al., 2017a) provides a high filtering speed but suffers from relatively high number of falsely accepted sequence pairs.
Our goal in this work is to significantly reduce the time spent on calculating the optimal alignment of short sequences and maintain high filtering accuracy. To this end, we introduce Shouji (Named after a traditional Japanese door that is designed to slide open http://www.aisf.or.jp/∼jaanus/deta/s/shouji.htm), a new, fast and very accurate pre-alignment filter. Shouji is based on two key ideas: (i) a new filtering algorithm that remarkably reduces the need for computationally expensive banded optimal alignment by rapidly excluding dissimilar sequences from the optimal alignment calculation. (ii) Judicious use of the parallelism-friendly architecture of modern FPGAs to greatly speed up this new filtering algorithm.
The contributions of this paper are as follows:
We introduce Shouji, a highly parallel and highly accurate pre-alignment filter, which uses a sliding search window approach to quickly identify dissimilar sequences without the need for computationally expensive alignment algorithms. We overcome the implementation limitations of MAGNET (Alser et al., 2017b). We build two hardware accelerator designs that adopt modern FPGA architectures to boost the performance of both Shouji and MAGNET.
We provide a comprehensive analysis of the run time and space complexity of Shouji and MAGNET algorithms. Shouji and MAGNET are asymptomatically inexpensive and run in linear time with respect to the sequence length and the edit distance threshold.
We demonstrate that Shouji and MAGNET significantly improve the accuracy of pre-alignment filtering by up to two and four orders of magnitude, respectively, compared to GateKeeper and SHD.
We demonstrate that our FPGA implementations of Shouji and MAGNET are two to three orders of magnitude faster than their CPU implementations. We demonstrate that integrating Shouji with five state-of-the-art aligners reduces the execution time of the sequence aligner by up to 18.8×.
2 Materials and methods
2.1 Overview
Our goal is to quickly reject dissimilar sequences with high accuracy such that we reduce the need for the computationally-costly alignment step. To this end, we propose the Shouji algorithm to achieve highly accurate filtering. Then, we accelerate Shouji by taking advantage of the parallelism of FPGAs to achieve fast filtering operations. The key filtering strategy of Shouji is inspired by the pigeonhole principle, which states that if E items are distributed into E + 1 boxes, then one or more boxes would remain empty. In the context of pre-alignment filtering, this principle provides the following key observation: if two sequences differ by E edits, then the two sequences should share at least a single common subsequence (i.e. free of edits) and at most E + 1 non-overlapping common subsequences, where E is the edit distance threshold. With the existence of at most E edits, the total length of these non-overlapping common subsequences should not be <m −E, where m is the sequence length. Shouji employs the pigeonhole principle to decide whether or not two sequences are potentially similar. Shouji finds all the non-overlapping subsequences that exist in both sequences. If the total length of these common subsequences <m−E, then there exist more edits than the allowed edit distance threshold, and hence Shouji rejects the two given sequences. Otherwise, Shouji accepts the two sequences. Next, we discuss the details of Shouji.
2.2 Shouji pre-alignment filter
Shouji identifies the dissimilar sequences, without calculating the optimal alignment, in three main steps. (i) The first step is to construct what we call a neighborhood map that visualizes the pairwise matches and mismatches between two sequences given an edit distance threshold of E characters. (ii) The second step is to find all the non-overlapping common subsequences in the neighborhood map using a sliding search window approach. (iii) The last step is to accept or reject the given sequence pairs based on the length of the found matches. If the length of the found matches is small, then Shouji rejects the input sequence pair.
2.2.1 Building the neighborhood map
We present in Figure 1 an example of a neighborhood map for two sequences, where a pattern P differs from a text T by three edits.
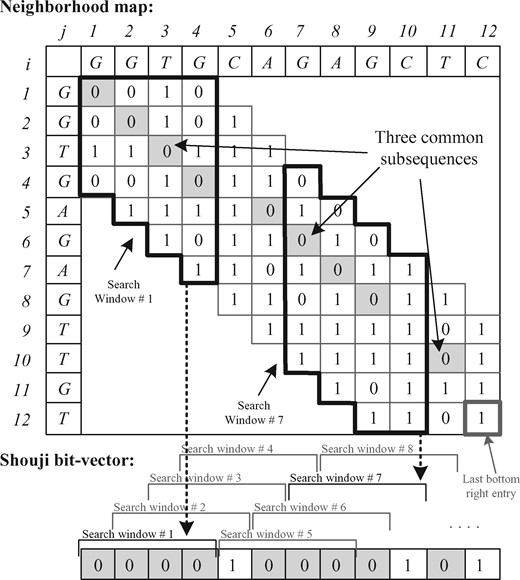
Neighborhood map (N) and the Shouji bit-vector, for text T=GGTGCAGAGCTC and pattern P=GGTGAGAGTTGT for E = 3. The three common subsequences (i.e. GGTG, AGAG and T) are highlighted in gray. We use a search window of size four columns (two examples of which are highlighted in black) with a step size of a single column. Shouji searches diagonally within each search window for the 4-bit vector that has the largest number of zeros. Once found, Shouji examines if the found 4-bit vector maximizes the number of zeros at the corresponding location of the 4-bit vector in the Shouji bit-vector. If so, then Shouji stores this 4-bit vector in the Shouji bit-vector at its corresponding location
The entry N[i, j] is set to zero if the ith character of the pattern matches the jth character of the text. Otherwise, it is set to one. The way we build our neighborhood map ensures that computing each of its entries is independent of every other, and thus the entire map can be computed all at once in a parallel fashion. Hence, our neighborhood map is well suited for highly parallel computing platforms (Alser et al., 2017a; Seshadri et al., 2017). Note that in sequence alignment algorithms, computing each entry of the dynamic programing matrix depends on the values of the immediate left, upper left and upper entries of its own. Different from ‘dot plot’ or ‘dot matrix’ (visual representation of the similarities between two closely similar genomic sequences) that is used in FASTA/FASTP (Lipman and Pearson, 1985), our neighborhood map computes only necessary diagonals near the main diagonal of the matrix (e.g. seven diagonals shown in Fig. 1).
2.2.2 Identifying the diagonally consecutive matches
The key goal of this step is to accurately find all the non-overlapping common subsequences shared between a pair of sequences. The accuracy of finding these subsequences is crucial for the overall filtering accuracy, as the filtering decision is made solely based on total subsequence length. With the existence of E edits, there are at most E + 1 non-overlapping common subsequences (based on the pigeonhole principle) shared between a pair of sequences. Each non-overlapping common subsequence is represented as a streak of diagonally consecutive zeros in the neighborhood map (as highlighted in yellow in Fig. 1). These streaks of diagonally consecutive zeros are distributed along the diagonals of the neighborhood map without any prior information about their length or number. One way of finding these common subsequences is to use a brute-force approach, which examines all the streaks of diagonally consecutive zeros that start at the first column and selects the streak that has the largest number of zeros as the first common subsequences. It then iterates over the remaining part of the neighborhood map to find the other common subsequences. However, this brute-force approach is infeasible for highly optimized hardware implementation as the search space is unknown at design time. Shouji overcomes this issue by dividing the neighborhood map into equal-size parts. We call each part a search window. Limiting the size of the search space from the entire neighborhood map to a search window has three key benefits. (i) It helps to provide a scalable architecture that can be implemented for any sequence length and edit distance threshold. (ii) Downsizing the search space into a reasonably small sub-matrix with a known dimension at design time limits the number of all possible permutations of each bit-vector to 2n, where n is the search window size. This reduces the size of the look-up tables (LUTs) required for an FPGA implementation and simplifies the overall design. (iii) Each search window is considered as a smaller sub-problem that can be solved independently and rapidly with high parallelism. Shouji uses a search window of four columns wide, as we illustrate in Figure 1. We need m search windows for processing two sequences, each of which is of length m characters. Each search window overlaps with its next neighboring search window by three columns. This ensures covering the entire neighborhood map and finding all the common subsequences regardless of their starting location. We select the width of each search window to be four columns to guarantee finding the shortest possible common subsequence, which is a single match located between two mismatches (i.e. ‘101’). However, we observe that the bit pattern ‘101’ is not always necessarily a part of the correct alignment (or the common subsequences). For example, the bit pattern ‘101’ exists once as a part of the correct alignment in Figure 1, but it also appears five times in other different locations that are not included in the correct alignment. To improve the accuracy of finding the diagonally consecutive matches, we increase the length of the diagonal vector to be examined to four bits. We also experimentally evaluate different search window sizes in Supplementary Materials, Section 6.1. We find that a search window size of four columns provides the highest filtering accuracy without falsely rejecting similar sequences.
Shouji finds the diagonally consecutive matches that are part of the common subsequences in the neighborhood map in two main steps. Step 1: for each search window, Shouji finds a 4-bit diagonal vector that has the largest number of zeros. Shouji greedily considers this vector as a part of the common subsequence as it has the least possible number of edits (i.e. 1’s). Finding always the maximum number of matches is necessary to avoid overestimating the actual number of edits and eventually preserving all similar sequences. Shouji achieves this step by comparing the 4 bits of each of the 2E + 1 diagonal vectors within a search window and selects the 4-bit vector that has the largest number of zeros. In the case where two 4-bit subsequences have the same number of zeros, Shouji breaks the ties by selecting the first one that has a leading zero. Then, Shouji slides the search window by a single column (i.e. step size =1 column) toward the last bottom right entry of the neighborhood map and repeats the previous computations. Thus, Shouji performs ‘Step 1’ m times using m search windows, where m is the sequence length. Step 2: the last step is to gather the results found for each search window (i.e. 4-bit vector that has the largest number of zeros) and construct back all the diagonally consecutive matches. For this purpose, Shouji maintains a Shouji bit-vector of length m that stores all the zeros found in the neighborhood map as we illustrate in Figure 1. For each sliding search window, Shouji examines if the selected 4-bit vector maximizes the number of zeros in the Shouji bit-vector at the same corresponding location. If so, Shouji stores the selected 4-bit vector in the Shouji bit-vector at the same corresponding location. This is necessary to avoid overestimating the number of edits between two given sequences. The common subsequences are represented as streaks of consecutive zeros in the Shouji bit-vector.
2.2.3 Filtering out dissimilar sequences
The last step of Shouji is to calculate the total number of edits (i.e. ones) in the Shouji bit-vector. Shouji examines if the total number of ones in the Shouji bit-vector >E. If so, Shouji excludes the two sequences from the optimal alignment calculation. Otherwise, Shouji considers the two sequences similar within the allowed edit distance threshold and allows their optimal alignment to be computed using optimal alignment algorithms. The Shouji bit-vector represents the differences between two sequences along the entire length of the sequence, m. However, Shouji is not limited to end-to-end edit distance calculation. Shouji is also able to provide edit distance calculation in local and glocal (semi-global) fashion. For example, achieving local edit distance calculation requires ignoring the ones that are located at the two ends of the Shouji bit-vector. We present an example of local edit distance between two sequences of different length in Supplementary Materials, Section 8. Achieving glocal edit distance calculation requires excluding the ones that are located at one of the two ends of the Shouji bit-vector from the total count of the ones in the Shouji bit-vector. This is important for correct pre-alignment filtering for global, local and glocal alignment algorithms. We provide the pseudocode of Shouji and discuss its computational complexity in Supplementary Materials, Section 6.2. We also present two examples of applying the Shouji filtering algorithm in Supplementary Materials, Section 8.
2.3 Accelerator architecture
Our second aim is to substantially accelerate Shouji, by leveraging the parallelism of FPGAs. In this section, we present our hardware accelerator that is designed to exploit the large amounts of parallelism offered by modern FPGA architectures (Aluru and Jammula, 2014; Herbordt et al., 2007; Trimberger, 2015). We then outline the implementation of Shouji to be used in our accelerator design. Figure 2 shows the hardware architecture of the accelerator. It contains a user-configurable number of filtering units. Each filtering unit provides pre-alignment filtering independently from other units. The workflow of the accelerator starts with transmitting the sequence pair to the FPGA through the fastest communication medium available on the FPGA board (i.e. PCIe). The sequence controller manages and provides the necessary input signals for each filtering unit in the accelerator. Each filtering unit requires two sequences of the same length and an edit distance threshold. The result controller gathers the output result (i.e. a single bit of value ‘1’ for similar sequences and ‘0’ for dissimilar sequences) of each filtering unit and transmits them back to the host side in the same order as their sequences are transmitted to the FPGAs.
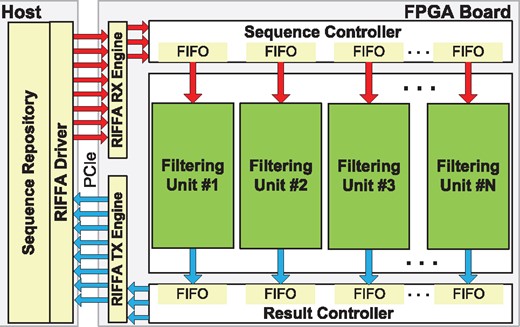
Overview of our hardware accelerator architecture. The filtering units can be replicated as many times as possible based on the resources available on the FPGA
The host-FPGA communication is achieved using RIFFA 2.2 (Jacobsen et al., 2015). To make the best use of the available resources in the FPGA chip, our algorithm utilizes the operations that are easily supported on an FPGA, such as bitwise operations, bit shifts and bit count. To build the neighborhood map on the FPGA, we use the observation that the main diagonal can be implemented using a bitwise XOR operation between the two given sequences. The upper E diagonals can be implemented by gradually shifting the pattern (P) to the right-hand direction and then performing bitwise XOR with the text (T). This allows each character of P to be compared with the right-hand neighbor characters (up to E characters) of its corresponding character of T. The lower E diagonals can be implemented in a way similar to the upper E diagonals, but here the shift operation is performed in the left-hand direction. This ensures that each character of P is compared with the left-hand neighbor characters (up to E characters) of its corresponding character of T.
We also build an efficient hardware architecture for each search window of the Shouji algorithm. It quickly finds the number of zeros in each 4-bit vector using a hardware look-up table that stores the 16 possible permutations of a 4-bit vector along with the number of zeros for each permutation. We present the block diagram of the search window architecture in Supplementary Materials, Section 6.3. Our hardware implementation of the Shouji filtering unit is independent of the specific FPGA-platform as it does not rely on any vendor-specific computing elements (e.g. intellectual property cores). However, each FPGA board has different resources and hardware capabilities that can directly or indirectly affect the performance and the data throughput of the design. The maximum data throughput of the design and the available FPGA resources determine the number of filtering units in the accelerator. Thus, if, e.g. the memory bandwidth is saturated, then increasing the number of filtering units would not improve performance.
3 Results
In this section, we evaluate (i) the filtering accuracy, (ii) the FPGA resource utilization, (iii) the execution time of Shouji, our hardware implementation of MAGNET (Alser et al., 2017b), GateKeeper (Alser et al., 2017a) and SHD (Xin et al., 2015), (iv) the benefits of the pre-alignment filters together with state-of-the-art aligners and (v) the benefits of Shouji together with state-of-the-art read mappers. As we mention in Section 1, MAGNET leads to a small number of falsely accepted sequence pairs but suffers from poor performance. We comprehensively explore this algorithm and provide an efficient and fast hardware implementation of MAGNET in Supplementary Materials, Section 7. We run all experiments using a 3.6 GHz Intel i7-3820 CPU with 8 GB RAM. We use a Xilinx Virtex 7 VC709 board (Xilinx, 2014) to implement our accelerator architecture (for both Shouji and MAGNET). We build the FPGA design using Vivado 2015.4 in synthesizable Verilog.
3.1 Dataset description
Our experimental evaluation uses 12 different real datasets. Each dataset contains 30 million real sequence pairs. We obtain three different read sets (ERR240727_1, SRR826460_1 and SRR826471_1) of the whole human genome that include three different read lengths (100, 150 and 250 bp). We download these three read sets from EMBL-ENA (www.ebi.ac.uk/ena). We map each read set to the human reference genome (GRCh37) using the mrFAST (Alkan et al., 2009) mapper. We obtain the human reference genome from the 1000 Genomes Project (1000 Genomes Project Consortium, 2012). For each read set, we use four different maximum numbers of allowed edits using the −e parameter of mrFAST to generate four real datasets. Each dataset contains the sequence pairs that are generated by the mrFAST mapper before the read alignment step. This enables us to measure the effectiveness of the filters using both aligned and unaligned sequences over a wide range of edit distance thresholds. We summarize the details of these 12 datasets in Supplementary Materials, Section 9. For the reader’s convenience, when referring to these datasets, we number them from 1 to 12 (e.g. set_1 to set_12). We use Edlib (Šošić and Šikić, 2017) to generate the ground truth edit distance value for each sequence pair.
3.2 Filtering accuracy
We evaluate the accuracy of a pre-alignment filter by computing its false accept rate and false reject rate. We first assess the false accept rate of Shouji, MAGNET (Alser et al., 2017b), SHD (Xin et al., 2015) and GateKeeper (Alser et al., 2017a) across different edit distance thresholds and datasets. The false accept rate is the ratio of the number of dissimilar sequences that are falsely accepted by the filter and the number of dissimilar sequences that are rejected by the optimal sequence alignment algorithm. We aim to minimize the false accept rate to maximize that number of dissimilar sequences that are eliminated. In Figure 3, we provide the false accept rate of the four filters across our 12 datasets and edit distance thresholds of 0–10% of the sequence length (we provide the exact values in Section 10 in Supplementary Materials).
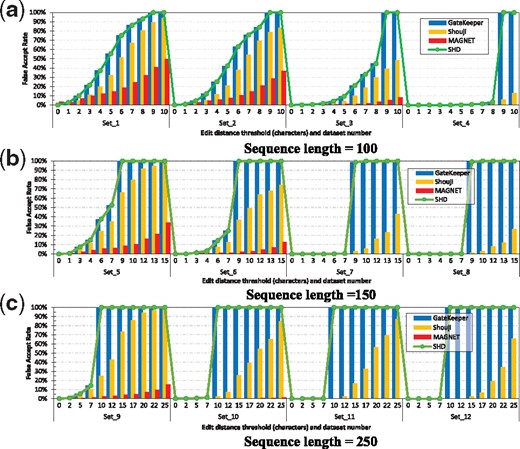
The false accept rate of Shouji, MAGNET, SHD and GateKeeper across 12 real datasets. We use a wide range of edit distance thresholds (0–10% of the sequence length) for sequence lengths of (a) 100, (b) 150 and (c) 250
Based on Figure 3, we make four key observations. (i) We observe that Shouji, MAGNET, SHD and GateKeeper are less accurate in examining the low-edit sequences (i.e. datasets 1, 2, 5, 6, 9 and 10) than the high-edit sequences (i.e. datasets 3, 4, 7, 8, 11 and 12).
(ii) SHD (Xin et al., 2015) and GateKeeper (Alser et al., 2017a) become ineffective for edit distance thresholds of >8% (E = 8), 5% (E = 7) and 3% (E = 7) for sequence lengths of 100, 150 and 250 characters, respectively. This causes them to examine each sequence pair unnecessarily twice (i.e. once by GateKeeper or SHD and once by the alignment algorithm). (iii) For high-edit datasets, Shouji provides up to 17.2, 73 and 467× (2.4, 2.7 and 38× for low-edit datasets) smaller false accept rate compared to GateKeeper and SHD for sequence lengths of 100, 150 and 250 characters, respectively. (iv) MAGNET shows up to 1577, 3550 and 25 552× lower false accept rates for high-edit datasets (3.5, 14.7 and 135× for low-edit datasets) compared to GateKeeper and SHD for sequence lengths of 100, 150 and 250 characters, respectively. MAGNET also shows up to 205, 951 and 16 760× lower false accept rates for high-edit datasets (2.7, 10 and 88× for low-edit datasets) over Shouji for sequence lengths of 100, 150 and 250 characters, respectively.
We conclude that Shouji and MAGNET (i) maintain a very low rate of falsely accepted dissimilar sequences and (ii) significantly improve the accuracy of pre-alignment filtering by up to two and four orders of magnitude, respectively, compared to GateKeeper and SHD.
Second, we assess the false reject rates of pre-alignment filters in Supplementary Materials, Section 10. We demonstrate that Shouji, SHD (Xin et al., 2015) and GateKeeper (Alser et al., 2017a) all have a 0% false reject rate. We also observe that MAGNET falsely rejects correct sequence pairs, which is unacceptable for a reliable filter. Hence, we conclude that Shouji is the most effective pre-alignment filter, with a low false accept rate and a zero false reject rate.
3.3 Data throughput and resource analysis
The operating frequency of our FPGA accelerator is 250 MHz. At this frequency, we observe a data throughput of nearly 3.3 GB/s, which corresponds to ∼13.3 billion bases per second. This nearly reaches the peak throughput of 3.64 GB/s provided by the RIFFA (Jacobsen et al., 2015) communication channel that feeds data into the FPGA using Gen3 4-lane PCIe. We examine the FPGA resource utilization of Shouji, MAGNET and GateKeeper (Alser et al., 2017a) filters. SHD (Xin et al., 2015) is implemented in C with Intel SSE instructions and cannot be directly implemented on an FPGA. We examine the FPGA resource utilization for two commonly used edit distance thresholds, 2 and 5% of the sequence length, as reported in (Ahmadi et al., 2012; Alser et al., 2017a; Hatem et al., 2013; Xin et al., 2015). The VC709 FPGA chip contains 433 200 slice LUTs (look-up tables) and 866 400 slice registers (flip-flops). Table 1 lists the FPGA resource utilization for a single filtering unit. We make three main observations. (i) The design for a single MAGNET filtering unit requires about 10.5 and 37.8% of the available LUTs for edit distance thresholds of 2 and 5, respectively. Hence, MAGNET can process 8 and 2 sequence pairs concurrently for edit distance thresholds of 2 and 5, respectively, without violating the timing constraints of our accelerator. (ii) The design for a single Shouji filtering unit requires about 15–21.9× fewer LUTs compared to MAGNET. This enables Shouji to achieve more parallelism over the MAGNET design as it can have 16 filtering units within the same FPGA chip. (iii) GateKeeper requires about 26.9–53× and 1.7–2.4× fewer LUTs compared to MAGNET and Shouji, respectively. GateKeeper can also examine 16 sequence pairs at the same time.
FPGA resource usage for a single filtering unit of Shouji, MAGNET and GateKeeper, for a sequence length of 100 and under different edit distance thresholds
| Filter | E | Single filtering unit | Max. no. of filtering units | |
|---|---|---|---|---|
| Slice LUT (%) | Slice register (%) | |||
| Shouji | 2 | 0.69 | 0.01 | 16 |
| 5 | 1.72 | 0.01 | 16 | |
| MAGNET | 2 | 10.50 | 0.8 | 8 |
| 5 | 37.80 | 2.30 | 2 | |
| GateKeeper | 2 | 0.39 | 0.01 | 16 |
| 5 | 0.71 | 0.01 | 16 | |
| Filter | E | Single filtering unit | Max. no. of filtering units | |
|---|---|---|---|---|
| Slice LUT (%) | Slice register (%) | |||
| Shouji | 2 | 0.69 | 0.01 | 16 |
| 5 | 1.72 | 0.01 | 16 | |
| MAGNET | 2 | 10.50 | 0.8 | 8 |
| 5 | 37.80 | 2.30 | 2 | |
| GateKeeper | 2 | 0.39 | 0.01 | 16 |
| 5 | 0.71 | 0.01 | 16 | |
Note: We highlight the best value in each column.
FPGA resource usage for a single filtering unit of Shouji, MAGNET and GateKeeper, for a sequence length of 100 and under different edit distance thresholds
| Filter | E | Single filtering unit | Max. no. of filtering units | |
|---|---|---|---|---|
| Slice LUT (%) | Slice register (%) | |||
| Shouji | 2 | 0.69 | 0.01 | 16 |
| 5 | 1.72 | 0.01 | 16 | |
| MAGNET | 2 | 10.50 | 0.8 | 8 |
| 5 | 37.80 | 2.30 | 2 | |
| GateKeeper | 2 | 0.39 | 0.01 | 16 |
| 5 | 0.71 | 0.01 | 16 | |
| Filter | E | Single filtering unit | Max. no. of filtering units | |
|---|---|---|---|---|
| Slice LUT (%) | Slice register (%) | |||
| Shouji | 2 | 0.69 | 0.01 | 16 |
| 5 | 1.72 | 0.01 | 16 | |
| MAGNET | 2 | 10.50 | 0.8 | 8 |
| 5 | 37.80 | 2.30 | 2 | |
| GateKeeper | 2 | 0.39 | 0.01 | 16 |
| 5 | 0.71 | 0.01 | 16 | |
Note: We highlight the best value in each column.
We conclude that the FPGA resource usage is correlated with the filtering accuracy. For example, the least accurate filter, GateKeeper, occupies the least FPGA resources. Yet, Shouji has very low FPGA resource usage.
3.4 Filtering speed
We analyze the execution time of MAGNET and Shouji compared to SHD (Xin et al., 2015) and GateKeeper (Alser et al., 2017a). We evaluate GateKeeper, MAGNET and Shouji using a single FPGA chip and run SHD using a single CPU core. SHD supports a sequence length of up to only 128 characters (due to the SIMD register size). To ensure as fair a comparison as possible, we allow SHD to divide the long sequences into batches of 128 characters, examine each batch individually, and then sum up the results. In Table 2, we provide the execution time of the four pre-alignment filters using 120 million sequence pairs under sequence lengths of 100 and 250 characters.
Execution time (in seconds) of FPGA-based GateKeeper, MAGNET, Shouji and CPU-based SHD under different edit distance thresholds and sequence lengths
| E | GateKeeper | MAGNET | Shouji | SHD |
|---|---|---|---|---|
| Sequence length=100 | ||||
| 2 | 2.89 a (0.18 b , 16 c ) | 2.89 (0.36, 8) | 2.89 (0.18, 16) | 60.33 |
| 5 | 2.89 (0.18, 16) | 2.89 (1.45, 2) | 2.89 (0.18, 16) | 67.92 |
| Sequence length=250 | ||||
| 5 | 5.78 (0.72, 8) | 5.78 (2.89d, 2) | 5.78 (0.72 d , 8) | 141.09 |
| 15 | 5.78 (0.72, 8) | 5.78 (5.78d, 1) | 5.78 (0.72 d , 8) | 163.82 |
| E | GateKeeper | MAGNET | Shouji | SHD |
|---|---|---|---|---|
| Sequence length=100 | ||||
| 2 | 2.89 a (0.18 b , 16 c ) | 2.89 (0.36, 8) | 2.89 (0.18, 16) | 60.33 |
| 5 | 2.89 (0.18, 16) | 2.89 (1.45, 2) | 2.89 (0.18, 16) | 67.92 |
| Sequence length=250 | ||||
| 5 | 5.78 (0.72, 8) | 5.78 (2.89d, 2) | 5.78 (0.72 d , 8) | 141.09 |
| 15 | 5.78 (0.72, 8) | 5.78 (5.78d, 1) | 5.78 (0.72 d , 8) | 163.82 |
Note: We use set_1 to set_4 for a sequence length of 100 and set_9 to set_12 for a sequence length of 250. We provide the performance results for both a single filtering unit and the maximum number of filtering units (in parentheses).
Execution time, in seconds, for a single filtering unit.
Execution time, in seconds, for maximum filtering units.
The number of filtering units.
Theoretical results based on the resource utilization and data throughput.
Execution time (in seconds) of FPGA-based GateKeeper, MAGNET, Shouji and CPU-based SHD under different edit distance thresholds and sequence lengths
| E | GateKeeper | MAGNET | Shouji | SHD |
|---|---|---|---|---|
| Sequence length=100 | ||||
| 2 | 2.89 a (0.18 b , 16 c ) | 2.89 (0.36, 8) | 2.89 (0.18, 16) | 60.33 |
| 5 | 2.89 (0.18, 16) | 2.89 (1.45, 2) | 2.89 (0.18, 16) | 67.92 |
| Sequence length=250 | ||||
| 5 | 5.78 (0.72, 8) | 5.78 (2.89d, 2) | 5.78 (0.72 d , 8) | 141.09 |
| 15 | 5.78 (0.72, 8) | 5.78 (5.78d, 1) | 5.78 (0.72 d , 8) | 163.82 |
| E | GateKeeper | MAGNET | Shouji | SHD |
|---|---|---|---|---|
| Sequence length=100 | ||||
| 2 | 2.89 a (0.18 b , 16 c ) | 2.89 (0.36, 8) | 2.89 (0.18, 16) | 60.33 |
| 5 | 2.89 (0.18, 16) | 2.89 (1.45, 2) | 2.89 (0.18, 16) | 67.92 |
| Sequence length=250 | ||||
| 5 | 5.78 (0.72, 8) | 5.78 (2.89d, 2) | 5.78 (0.72 d , 8) | 141.09 |
| 15 | 5.78 (0.72, 8) | 5.78 (5.78d, 1) | 5.78 (0.72 d , 8) | 163.82 |
Note: We use set_1 to set_4 for a sequence length of 100 and set_9 to set_12 for a sequence length of 250. We provide the performance results for both a single filtering unit and the maximum number of filtering units (in parentheses).
Execution time, in seconds, for a single filtering unit.
Execution time, in seconds, for maximum filtering units.
The number of filtering units.
Theoretical results based on the resource utilization and data throughput.
We make four key observations. (i) Shouji’s execution time is as low as that of GateKeeper (Alser et al., 2017a), and 2–8× lower than that of MAGNET. This observation is in accord with our expectation and can be explained by the fact that MAGNET has more resource overhead that limits the number of filtering units on an FPGA. Yet Shouji is up to two orders of magnitude more accurate than GateKeeper (as we show earlier in Section 3.2). (ii) Shouji is up to 28 and 335× faster than SHD using one and 16 filtering units, respectively. (iii) MAGNET is up to 28 and 167.5× faster than SHD using one and eight filtering units, respectively. As we present in Supplementary Materials, Section 12, the hardware-accelerated versions of Shouji and MAGNET provide up to three orders of magnitude of speedup over their functionally equivalent CPU implementations.
We conclude that Shouji is extremely fast and accurate. Shouji’s performance also scales very well over a wide range of both edit distance thresholds and sequence lengths.
3.5 Effects of pre-alignment filtering on sequence alignment
We analyze the benefits of integrating our proposed pre-alignment filter (and other filters) with state-of-the-art aligners. Table 3 presents the effect of different pre-alignment filters on the overall alignment time. We select five best-performing aligners, each of which is designed for a different type of computing platform. We use a total of 120 million real sequence pairs from our previously described four datasets (set_1 to set_4) in this analysis. We evaluate the actual execution time of Edlib (Šošić and Šikić, 2017) and Parasail (Daily, 2016) on our machine. However, FPGASW (Fei et al., 2018), CUDASW++ 3.0 (Liu et al., 2013) and GSWABE (Liu and Schmidt, 2015) are not open-source and not available to us. Therefore, we scale the reported number of computed entries of the dynamic programming matrix in a second (i.e. GCUPS) as follows: 120 000 000/(GCUPS/1002). We make three key observations. (i) The execution time of Edlib (Šošić and Šikić, 2017) reduces by up to 18.8, 16.5, 13.9 and 5.2× after the addition of Shouji, MAGNET, GateKeeper and SHD, respectively, as a pre-alignment filtering step. We also observe a very similar trend for Parasail (Daily, 2016) combined with each of the four pre-alignment filters. (ii) Aligners designed for FPGAs and GPUs follow a different trend than that we observe in the CPU aligners. We observe that FPGASW (Fei et al., 2018), CUDASW++ 3.0 (Liu et al., 2013) and GSWABE (Liu and Schmidt, 2015) are faster alone than with SHD (Xin et al., 2015) incorporated as the pre-alignment filtering step. Shouji, MAGNET and GateKeeper (Alser et al., 2017a) still significantly reduce the overall execution time of both FPGA and GPU based aligners. Shouji reduces the overall alignment time of FPGASW (Fei et al., 2018), CUDASW++ 3.0 (Liu et al., 2013) and GSWABE (Liu and Schmidt, 2015) by factors of up to 14.5, 14.2 and 17.9×, respectively. This is up to 1.35, 1.4 and 85× more than the effect of MAGNET, GateKeeper and SHD on the end-to-end alignment time.
End-to-end execution time (in seconds) for several state-of-the-art sequence alignment algorithms, with and without pre-alignment filters (Shouji, MAGNET, GateKeeper and SHD) and across different edit distance thresholds
| E | Edlib | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
|---|---|---|---|---|---|
| 2 | 506.66 | 26.86 | 30.69 | 36.39 | 96.54 |
| 5 | 632.95 | 147.20 | 106.80 | 208.77 | 276.51 |
| E | Parasail | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 1310.96 | 69.21 | 78.83 | 93.87 | 154.02 |
| 5 | 2044.58 | 475.08 | 341.77 | 673.99 | 741.73 |
| E | FPGASW | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 11.33 | 0.78 | 1.04 | 0.99 | 61.14 |
| 5 | 11.33 | 2.81 | 3.34 | 3.91 | 71.65 |
| E | CUDASW++ 3.0 | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 10.08 | 0.71 | 0.96 | 0.90 | 61.05 |
| 5 | 10.08 | 2.52 | 3.13 | 3.50 | 71.24 |
| E | GSWABE | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 61.86 | 3.44 | 4.06 | 4.60 | 64.75 |
| 5 | 61.86 | 14.55 | 11.75 | 20.57 | 88.31 |
| E | Edlib | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
|---|---|---|---|---|---|
| 2 | 506.66 | 26.86 | 30.69 | 36.39 | 96.54 |
| 5 | 632.95 | 147.20 | 106.80 | 208.77 | 276.51 |
| E | Parasail | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 1310.96 | 69.21 | 78.83 | 93.87 | 154.02 |
| 5 | 2044.58 | 475.08 | 341.77 | 673.99 | 741.73 |
| E | FPGASW | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 11.33 | 0.78 | 1.04 | 0.99 | 61.14 |
| 5 | 11.33 | 2.81 | 3.34 | 3.91 | 71.65 |
| E | CUDASW++ 3.0 | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 10.08 | 0.71 | 0.96 | 0.90 | 61.05 |
| 5 | 10.08 | 2.52 | 3.13 | 3.50 | 71.24 |
| E | GSWABE | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 61.86 | 3.44 | 4.06 | 4.60 | 64.75 |
| 5 | 61.86 | 14.55 | 11.75 | 20.57 | 88.31 |
We highlight the best value in each row.
End-to-end execution time (in seconds) for several state-of-the-art sequence alignment algorithms, with and without pre-alignment filters (Shouji, MAGNET, GateKeeper and SHD) and across different edit distance thresholds
| E | Edlib | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
|---|---|---|---|---|---|
| 2 | 506.66 | 26.86 | 30.69 | 36.39 | 96.54 |
| 5 | 632.95 | 147.20 | 106.80 | 208.77 | 276.51 |
| E | Parasail | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 1310.96 | 69.21 | 78.83 | 93.87 | 154.02 |
| 5 | 2044.58 | 475.08 | 341.77 | 673.99 | 741.73 |
| E | FPGASW | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 11.33 | 0.78 | 1.04 | 0.99 | 61.14 |
| 5 | 11.33 | 2.81 | 3.34 | 3.91 | 71.65 |
| E | CUDASW++ 3.0 | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 10.08 | 0.71 | 0.96 | 0.90 | 61.05 |
| 5 | 10.08 | 2.52 | 3.13 | 3.50 | 71.24 |
| E | GSWABE | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 61.86 | 3.44 | 4.06 | 4.60 | 64.75 |
| 5 | 61.86 | 14.55 | 11.75 | 20.57 | 88.31 |
| E | Edlib | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
|---|---|---|---|---|---|
| 2 | 506.66 | 26.86 | 30.69 | 36.39 | 96.54 |
| 5 | 632.95 | 147.20 | 106.80 | 208.77 | 276.51 |
| E | Parasail | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 1310.96 | 69.21 | 78.83 | 93.87 | 154.02 |
| 5 | 2044.58 | 475.08 | 341.77 | 673.99 | 741.73 |
| E | FPGASW | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 11.33 | 0.78 | 1.04 | 0.99 | 61.14 |
| 5 | 11.33 | 2.81 | 3.34 | 3.91 | 71.65 |
| E | CUDASW++ 3.0 | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 10.08 | 0.71 | 0.96 | 0.90 | 61.05 |
| 5 | 10.08 | 2.52 | 3.13 | 3.50 | 71.24 |
| E | GSWABE | w/ Shouji | w/ MAGNET | w/ GateKeeper | w/ SHD |
| 2 | 61.86 | 3.44 | 4.06 | 4.60 | 64.75 |
| 5 | 61.86 | 14.55 | 11.75 | 20.57 | 88.31 |
We highlight the best value in each row.
(iii) We observe that if the execution time of the aligner is much larger than that of the pre-alignment filter (which is the case for Edlib, Parasail and GSWABE for E = 5 characters), then MAGNET provides up to 1.3× more end-to-end speedup over Shouji. This is expected as MAGNET produces a smaller false accept rate compared to Shouji. However, unlike MAGNET, Shouji provides a 0% false reject rate. We conclude that among the four pre-alignment filters, Shouji is the best-performing pre-alignment filter in terms of both speed and accuracy. Integrating Shouji with an aligner leads to strongly positive benefits and reduces the aligner’s total execution time by up to 18.8×.
3.6 Effects of pre-alignment filtering on the read mapper
After confirming the benefits of integrating Shouji with sequence alignment algorithms, we now evaluate the overall benefits of integrating Shouji with the mrFAST (v. 2.6.1) mapper (Alkan et al., 2009) and BWA-MEM (Li, 2013). Table 4 summarizes the effect of Shouji on the overall mapping time, when all reads from ERR240727_1 (100 bp) are mapped to GRCh37 with an edit distance threshold of 2 and 5%. We also provide the total execution time breakdown in Supplementary Table S15. We make two observations. (i) The mapping time of mrFAST reduces by a factor of up to five after adding Shouji as the pre-alignment step. (ii) Integrating Shouji with BWA-MEM, without optimizing the mapper, shows less benefit than integrating Shouji with mrFAST (up to 1.07× reduction in the overall mapping time). This is due to the fact that BWA-MEM generates a low number of pairs that require verification using the read aligner. We believe by changing the mapper to work better with Shouji, we can achieve larger speedups. We leave this for future work.
Overall mrFAST and BWA-MEM mapping time (in seconds) with and without Shouji, for an edit distance threshold of 2 and 5%
| E | # pairs to be verified | # pairs rejected by Shouji | Map. time w/o Shouji (s) | Mapping time w/ Shouji | |
|---|---|---|---|---|---|
| mrFAST | 2 | 40 859 970 | 30 679 795 | 242.1 | 195.4s (1.2×) |
| 5 | 874 403 170 | 764 688 027 | 2532 | 504.6s (5.0×) | |
| BWA-MEM | 2 | 653 543 | 585 036 | 668.1 | 626.9s (1.07×) |
| 2a | 8 209 193 | 7 847 125 | 670.1 | 625.8s (1.07×) | |
| 5 | 660 901 | 593 247 | 695.1 | 655.8s (1.06×) | |
| 5a | 8 542 937 | 8 186 550 | 696.1 | 652.7s (1.07×) |
| E | # pairs to be verified | # pairs rejected by Shouji | Map. time w/o Shouji (s) | Mapping time w/ Shouji | |
|---|---|---|---|---|---|
| mrFAST | 2 | 40 859 970 | 30 679 795 | 242.1 | 195.4s (1.2×) |
| 5 | 874 403 170 | 764 688 027 | 2532 | 504.6s (5.0×) | |
| BWA-MEM | 2 | 653 543 | 585 036 | 668.1 | 626.9s (1.07×) |
| 2a | 8 209 193 | 7 847 125 | 670.1 | 625.8s (1.07×) | |
| 5 | 660 901 | 593 247 | 695.1 | 655.8s (1.06×) | |
| 5a | 8 542 937 | 8 186 550 | 696.1 | 652.7s (1.07×) |
We configure BWA-MEM to report all secondary alignments using −a.
Overall mrFAST and BWA-MEM mapping time (in seconds) with and without Shouji, for an edit distance threshold of 2 and 5%
| E | # pairs to be verified | # pairs rejected by Shouji | Map. time w/o Shouji (s) | Mapping time w/ Shouji | |
|---|---|---|---|---|---|
| mrFAST | 2 | 40 859 970 | 30 679 795 | 242.1 | 195.4s (1.2×) |
| 5 | 874 403 170 | 764 688 027 | 2532 | 504.6s (5.0×) | |
| BWA-MEM | 2 | 653 543 | 585 036 | 668.1 | 626.9s (1.07×) |
| 2a | 8 209 193 | 7 847 125 | 670.1 | 625.8s (1.07×) | |
| 5 | 660 901 | 593 247 | 695.1 | 655.8s (1.06×) | |
| 5a | 8 542 937 | 8 186 550 | 696.1 | 652.7s (1.07×) |
| E | # pairs to be verified | # pairs rejected by Shouji | Map. time w/o Shouji (s) | Mapping time w/ Shouji | |
|---|---|---|---|---|---|
| mrFAST | 2 | 40 859 970 | 30 679 795 | 242.1 | 195.4s (1.2×) |
| 5 | 874 403 170 | 764 688 027 | 2532 | 504.6s (5.0×) | |
| BWA-MEM | 2 | 653 543 | 585 036 | 668.1 | 626.9s (1.07×) |
| 2a | 8 209 193 | 7 847 125 | 670.1 | 625.8s (1.07×) | |
| 5 | 660 901 | 593 247 | 695.1 | 655.8s (1.06×) | |
| 5a | 8 542 937 | 8 186 550 | 696.1 | 652.7s (1.07×) |
We configure BWA-MEM to report all secondary alignments using −a.
4 Discussion and future work
We demonstrate that the concept of pre-alignment filtering provides substantial benefits to the existing and future sequence alignment algorithms. Accelerated sequence aligners that offer different strengths and features are frequently introduced. Many of these efforts either simplify the scoring function, or only take into account accelerating the computation of the dynamic programming matrix without supporting the backtracking step. Shouji offers the ability to make the best use of existing aligners without sacrificing any of their capabilities, as it does not modify or replace the alignment step. As such, we hope that it catalyzes the adoption of specialized pre-alignment accelerators in genome sequence analysis. However, the use of specialized hardware chips may discourage users who are not necessarily fluent in FPGAs. This concern can be alleviated in at least two ways. First, the Shouji accelerator can be integrated more closely inside the sequencing machines to perform real-time pre-alignment filtering concurrently with sequencing (Lindner et al., 2016). This allows a significant reduction in total genome analysis time. Second, cloud computing offers access to a large number of advanced FPGA chips that can be used concurrently via a simple user-friendly interface. However, such a scenario requires the development of privacy-preserving pre-alignment filters due to privacy and legal concerns (Salinas and Li, 2017). Our next efforts will focus on exploring privacy-preserving real-time pre-alignment filtering.
Another potential target of our research is to explore the possibility of accelerating optimal alignment calculations for longer sequences (few tens of thousands of characters) (Senol et al., 2018) using pre-alignment filtering. Longer sequences pose two challenges. First, we need to transfer more data to the FPGA chip to be able process a single pair of sequences which is mainly limited by the data transfer rate of the communication link (i.e. PCIe). Second, typical edit distance threshold used for sequence alignment is 5% of the sequence length. For considerably long sequences, edit distance threshold is around few hundreds of characters. For a large edit distance threshold, each character of a given sequence is compared to a large number of neighboring characters of the other given sequence. This makes the short matches (e.g. a single zero or two consecutive zeros) to occur more frequently in the diagonal vectors, which would negatively affect the accuracy of Shouji. We will investigate this effect and explore new pre-alignment filtering approaches for the sequencing data produced by third-generation sequence machines.
5 Conclusion
In this work, we propose Shouji, a highly parallel and accurate pre-alignment filtering algorithm accelerated on a specialized hardware platform. The key idea of Shouji is to rapidly and accurately eliminate dissimilar sequences without calculating banded optimal alignment. Our hardware-accelerated version of Shouji provides, on average, three orders of magnitude speedup over its functionally equivalent CPU implementation. Shouji improves the accuracy of pre-alignment filtering by up to two orders of magnitude compared to the best-performing existing pre-alignment filter, GateKeeper. The addition of Shouji as a pre-alignment step significantly reduces the alignment time of state-of-the-art aligners by up to 18.8×, leading to the fastest alignment mechanism that we know of.
Funding
This work was supported in part by the National Institutes of Health grant [HG006004 to O.M. and C.A.]; and the EMBO Installation grant [IG-2521 to C.A.]. M.A. is supported in part by the HiPEAC collaboration grant and TUBITAK-2215 graduate fellowship from the Scientific and Technological Research Council of Turkey.
Conflict of Interest: none declared.
Acknowledgements
We thank Tuan Duy Anh Nguyen for his valuable comments on the hardware design.
References
Xilinx (


{kind=link}
{kind=link}
{kind=link}