





Abstract
Tandem DNA repeats can be sequenced with long-read technologies, but cannot be accurately deciphered due to the lack of computational tools taking high error rates of these technologies into account. Here we introduce Noise-Cancelling Repeat Finder (NCRF) to uncover putative tandem repeats of specified motifs in noisy long reads produced by Pacific Biosciences and Oxford Nanopore sequencers. Using simulations, we validated the use of NCRF to locate tandem repeats with motifs of various lengths and demonstrated its superior performance as compared to two alternative tools. Using real human whole-genome sequencing data, NCRF identified long arrays of the (AATGG)n repeat involved in heat shock stress response.
NCRF is implemented in C, supported by several python scripts, and is available in bioconda and at https://github.com/makovalab-psu/NoiseCancellingRepeatFinder.
Supplementary data are available at Bioinformatics online.
1 Introduction
Long tandem repeat (TR) arrays are associated with heterochromatin and play critical roles in the human genome. For instance, (TTAGGG)n TRs protect telomeres (Blackburn and Gall, 1978), (AATGG)n repeats are implicated in heat shock response (Goenka et al., 2016), and the lengths of heterochromatin-associated TRs differ across populations (Altemose et al., 2014; Wevrick and Willard, 1989) and change with aging and environmental exposure (Goenka et al., 2016; Zhang et al., 2015). Despite these important features of TRs, their length variation has been understudied due to a lack of experimental and computational techniques able to capture their full length.
Long TRs cannot be studied with short sequencing reads, but can be profiled with long-read technologies (Pacific Biosciences, or PacBio, and Oxford Nanopore, or Nanopore). However, they are difficult to decipher because such technologies have distinctive error profiles (see below). Moreover, they are often absent from reference genomes and assemblies (Peona et al., 2018). To our knowledge, no tool currently exists to identify TR arrays in long, error-prone reads. Tools solving similar problems, primarily developed to work with short reads or assembled genomes, have limitations when applied to this use case (Lower et al., 2018). Some fail to consider unequal rates of insertions versus deletions [e.g. Tandem Repeats Finder, or TRF (Benson, 1999)]; others do not permit high sequencing error rates (e.g. short read mappers). General purpose aligners, e.g. Minimap2 (Li, 2018), even with parameterizations for long-read sequencing technologies, are not designed to find TRs.
To address the shortcomings of existing tools in identifying user-specified TR arrays directly from error-prone long sequencing reads, we developed Noise-Cancelling Repeat Finder (NCRF). NCRF supports high and unequal rates of short insertions and deletions observed in long-read sequencing data. As a result, its performance is superior to alternative tools.
2 Developing NCRF
The aligner at the core of NCRF finds alignments of a given motif to a segment of a given DNA sequence, with the motif repeated as often as needed. It is a Smith–Waterman aligner (Smith and Waterman, 1981) with affine gap penalties. It makes use of a typical Dynamic Programming matrix with a row for each nucleotide in a single copy of the motif and a column for each nucleotide in the sequence, allowing for wraparound from the end to the beginning of the motif (Supplementary Note S1A). Typically, the alignment core utilizes a score for matches and penalties for mismatches and indels; but we allow different penalties for insertions and deletions because sequencing technologies can be biased as to which type of indel they introduce. Thus, technology-specific scoring parameters are tuned to observed sequencing error profiles (Supplementary Notes S1D, S2). The dynamic programming recurrence is further modified to support a high prevalence of short indels (Supplementary Note S1A). A finishing step filters out and discards alignment pieces with a high density of mismatches and indels, retaining only high-quality alignments (Supplementary Note S1B).
Alignments identify intervals that putatively align to perfectly repeated copies of a motif. However, segments containing a mix of motif variants, or a similar motif, may also be reported. Such mixes are consistent with known evolutionary signatures of heterochromatic repeats (Plohl et al., 2008). An optional consensus filtering step eliminates TR arrays lacking a single dominant motif. Intervals reported for more than one motif can be identified with an optional overlap-detection step, see Supplementary Note S1 and Supplementary Figure S1 for details.
3 Analysis of simulated reads and tool comparison
We simulated PacBio and Nanopore sequencing reads for a mock genome mimicking the presence of long repeat arrays in the human reference genome (Supplementary Note S3). NCRF discovered 99% and 91% of the specified TRs in PacBio and Nanopore reads, respectively (Fig. 1A and Supplementary Table S2). In comparison, TRF discovered only 72% and 33%, while Minimap2 60% and 63%, for PacBio and Nanopore reads, respectively. False discovery rate (FDR) was much higher for NCRF than for TRF and Minimap2. Thus, we introduced the optional consensus filtering step in NCRF, reducing the FDR to below 1%, while still outperforming both TRF and Minimap2 in true positive rate (TPR). For the remainder of this section, we refer only to consensus-filtered results.
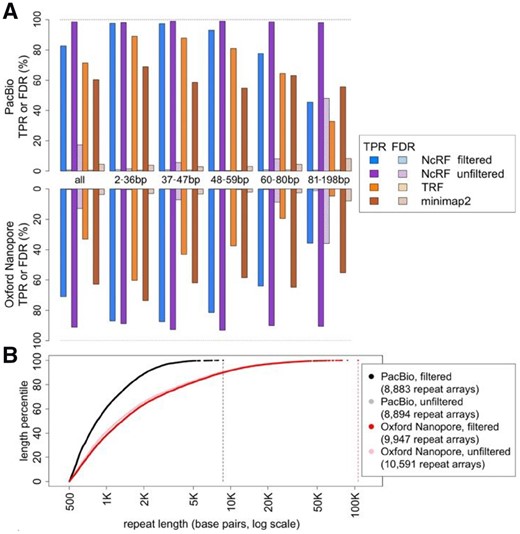
(A) Performance of NCRF, TRF, and Minimap2 on simulated PacBio (upper panel) and Nanopore (lower panel) reads, binned by motif lengths. Solid bars are TPRs, crosshatched bars are FDRs. All 847 arrays totaled 822 kb, with 197 arrays and 170 kb for lengths 2–36 bp, 156/158 kb for 37–47 bp, 158/173 kb for 48–59 bp, 172/162 kb for 60–80 bp and 164/160 kb for 81–198 bp. (B) Observed lengths of (AATGG)n arrays (with and without consensus filtering) in PacBio and Nanopore reads. Reads were subsampled to a similar length distribution of 16.5 Gb (Supplementary Note S5). Filtered and unfiltered results for PacBio are very similar
Further, we studied how the performance of all three tools was affected by the motif length. For this analysis, we divided mock repeat arrays into five bins by motif length (2–36 bp, 37–47 bp, 48–59 bp, 60–80 bp and 81–198 bp), each bin having ∼20% of the total repeat bases in the mock genome. In the two shortest bins, NCRF had TPRs of 97% for Pacbio and 87% for Nanopore. This rate decreased as motifs grew longer—to 93% and 81%, respectively, for the middle bin, to 78% and 64% for the fourth bin, and to 45% and 36% for the longest bin. The same trend was observed for TRF, with TPR decreasing for longer bins. In all bins NCRF’s TPR was higher than TRF’s. For PacBio, NCRF’s TPR was between 8% and 13% higher than TRF; for Nanopore, it was 27% to 45% higher. In contrast, TPR for Minimap2 fluctuated, apparently independent of the motif length. Still, NCRF had higher TPR for the short and middle bins, as well as the fourth bin for PacBio. Comparing FDRs, NCRF’s FDR was below 1.2% across the board. TRF had better (lower) FDR in all bins but one; however this minor advantage (typically <0.2%) pales in comparison to NCRF’s gain in TPR. Minimap2’s FDR was worse than both NCRF and TRF in all bins. Surprisingly, both TRF and Minimap2 occasionally reported overlapping intervals for the same motif (Supplementary Table S2). Several other tools were considered for this evaluation but rejected after preliminary investigation (Supplementary Note S4).
4 Applying NCRF to real sequencing data
Lastly, we applied NCRF to investigate perfect repeats of (AATGG)n in publicly available PacBio and Nanopore sequenced data (Jain et al., 2018; Zook et al., 2016) generated for the same individual, subsampled to a 16.5 Gb common read length distribution (Supplementary Note S5). Searching for >500-bp repeats of (AATGG)n, NCRF identified 8883 repeats in PacBio covering 9.8 Mb; averaging 0.6 bp per kb sequenced (Fig. 1B). 9947 repeats covering 35.6 Mb were found in Nanopore; 2.2 bp per kb sequenced. Additional applications of NCRF to real sequencing data, as well as potential reasons behind differences in density between technologies, are presented in Cechova et al. (2018).
5 Conclusions
To our knowledge, NCRF is the first tool designed specifically to identify TR arrays in noisy and reference-free sequencing data, accounting for the unique characteristics of the long-read technologies. We anticipate NCRF will accelerate research of heterochromatin-associated TR arrays and will aid in unraveling their functions in the genome.
Funding
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No, R01GM130691. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding was also provided by the Eberly College of Sciences, The Huck Institute of Life Sciences, and the Institute for CyberScience, at Penn State, as well as under grants from the Pennsylvania Department of Health using Tobacco Settlement and CURE Funds.
Conflict of Interest: none declared.
References


{kind=link}