





Abstract
Summary: Computational prediction of protein structure from sequence is broadly viewed as a foundational problem of biochemistry and one of the most difficult challenges in bioinformatics. Once every two years the Critical Assessment of protein Structure Prediction (CASP) experiments are held to assess the state of the art in the field in a blind fashion, by presenting predictor groups with protein sequences whose structures have been solved but have not yet been made publicly available. The first CASP was organized in 1994, and the latest, CASP13, took place last December, when for the first time the industrial laboratory DeepMind entered the competition. DeepMind's entry, AlphaFold, placed first in the Free Modeling (FM) category, which assesses methods on their ability to predict novel protein folds (the Zhang group placed first in the Template-Based Modeling (TBM) category, which assess methods on predicting proteins whose folds are related to ones already in the Protein Data Bank.) DeepMind's success generated significant public interest. Their approach builds on two ideas developed in the academic community during the preceding decade: (i) the use of co-evolutionary analysis to map residue co-variation in protein sequence to physical contact in protein structure, and (ii) the application of deep neural networks to robustly identify patterns in protein sequence and co-evolutionary couplings and convert them into contact maps. In this Letter, we contextualize the significance of DeepMind's entry within the broader history of CASP, relate AlphaFold's methodological advances to prior work, and speculate on the future of this important problem.
1 Significance
Progress in Free Modeling (FM) prediction in Critical Assessment of protein Structure Prediction (CASP) has historically ebbed and flowed, with a 10-year period of relative stagnation finally broken by the advances seen at CASP11 and 12, which were driven by the advent of co-evolution methods (Moult et al., 2016, 2018; Ovchinnikov et al., 2016; Schaarschmidt et al., 2018; Zhang et al., 2018) and the application of deep convolutional neural networks (Wang et al., 2017). The progress at CASP13 corresponds to roughly twice the recent rate of advance [measured in mean ΔGDT_TS from CASP10 to CASP12—GDT_TS is a measure of prediction accuracy ranging from 0 to 100, with 100 being perfect (Zemla et al., 1999)]. This can be observed not only in the CASP-over-CASP improvement, but also in the size of the gap between AlphaFold and the second best performer at CAPS13, which is unusually large by CASP standards (Fig. 1). Even when excluding AlphaFold, CASP13 shows further progress due to the widespread adoption of deep learning and the continued exploitation of co-evolutionary information in protein structure prediction (de Oliveira and Deane, 2017). Taken together these observations indicate substantial progress both by the whole field and by AlphaFold in particular.
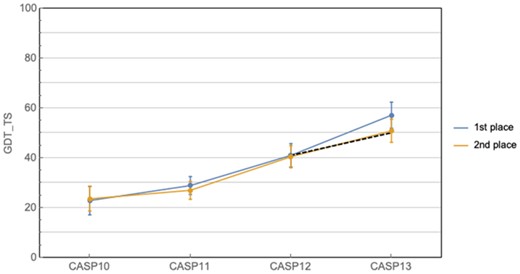
Historical CASP performance in prediction of gross protein topology. Curves show the best and second best predictors at each CASP, while the dashed line shows the expected improvement at CASP13 given the average rate of improvement from CASP10 to 12. Ranking is based on CASP assessor’s formula, and does not always coincide with highest mean GDT_TS (e.g. CASP10). Error bars correspond to 95% confidence intervals
Nonetheless, the problem remains far from solved, particularly for practical applications. GDT_TS measures gross topology, which is of inherent biological interest, but does not necessarily result in structures useful in drug discovery or molecular biology applications. An alternate metric, GDT_HA, provides a more stringent assessment of structural accuracy (Read and Chavali, 2007). Figure 2 plots the GDT_HA scores of the top two performers for the last four CASPs. While substantial progress can be discerned, the distance to perfect predictions remains sizeable. In addition, both metrics measure global goodness of fit, which can mask significant local deviations. Local accuracy corresponding to, for example, the coordination of atoms in an active site or the localized change of conformation due to a mutation, can be the most important aspect of a predicted structure when answering broader biological questions.
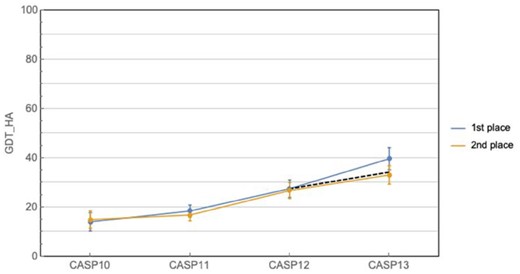
Historical CASP performance in prediction of fine-grained protein topology. Curves show the best and second best predictors at each CASP, while the dashed line shows the expected improvement at CASP13 given the average rate of improvement from CASP10 to 12. Ranking is based on CASP assessor’s formula, and does not always coincide with highest mean GDT_HA (e.g. CASP10). Error bars correspond to 95% confidence intervals
It remains the case however that AlphaFold represents an anomalous leap in protein structure prediction and portends favorably for the future. In particular, if the AlphaFold-adjusted trend in Figure 1 were to continue, then it is conceivable that in ∼5 years’ time we will begin to expect predicted structures with a mean GDT_TS of ∼85%, which would arguably correspond to a solution of the gross topology problem. Whether the trend will continue remains to be seen. The exponential increase in the number of sequenced proteins virtually ensures that improvements will be had even without new methodological developments. However, for the more general problem of predicting arbitrary protein structures from an individual amino acid sequence, including designed ones, new conceptual breakthroughs will almost certainly be required to obtain further progress.
2 Prior work
AlphaFold is a co-evolution-dependent method building on the groundwork laid by several research groups over the preceding decade. Co-evolution methods work by first constructing a multiple sequence alignment (MSA) of proteins homologous to the protein of interest. Such MSAs must be large, often comprising 105–106 sequences, and evolutionarily diverse (Tetchner et al., 2014). The so-called evolutionary couplings are then extracted from the MSA by detecting residues that co-evolve, i.e. that have mutated over evolutionary timeframes in response to other mutations, thereby suggesting physical proximity in space. The foundational methodology behind this approach was developed two decades ago (Lapedes et al., 1999), but was originally only validated in simulation as large protein sequence families were not yet available. The first set of such approaches to be applied effectively to real proteins came after the exponential increase in availability of protein sequences (Jones et al., 2012; Kamisetty et al., 2013; Marks et al., 2011; Weigt et al., 2009). These approaches predicted binary contact matrices from MSAs, i.e. whether two residues are ‘in contact’ or not (typically defined as having Cβ atoms within <8 Å), and fed that information to geometric constraint satisfaction methods such as CNS (Brünger et al., 1998) to fold the protein and obtain its 3D coordinates. This first generation of methods was a significant breakthrough, and ushered in the new era of protein structure prediction.
An important if expected development was the coupling of binary contacts with more advanced folding pipelines, such as Rosetta (Leaver-Fay et al., 2011) and I-Tasser (Yang et al., 2015), which resulted in better accuracy and constituted the state of the art in the FM category until the beginning of CASP12. The next major advance came from applying convolutional networks (LeCun et al., 2015) and deep residual networks (He et al., 2015; Srivastava et al., 2015) to integrate information globally across the entire matrix of raw evolutionary couplings to obtain more accurate contacts (Liu et al., 2018; Wang et al., 2017). This led to some of the advances seen at CASP12, although ultimately the best performing group at CASP12 did not make extensive use of deep learning [convolutional neural networks made a significant impact on contact prediction at CASP12, but the leading method was not yet fully implemented to have an impact on structure prediction (Wang et al., 2017)].
During the lead up to CASP13, one group published a modification to their method, RaptorX (Xu, 2018), that proved highly consequential. As before, RaptorX takes MSAs as inputs, but instead of predicting binary contacts, it predicts discrete distances. Specifically, RaptorX predicts probabilities over discretized spatial ranges (e.g. 10% probability for 4–4.5 Å), then uses the mean and variance of the predicted distribution to calculate lower and upper bounds for atom–atom distances. These bounds are then fed to CNS to fold the protein. RaptorX showed promise on a subset of CASP13 targets, with its seemingly simple change having a surprisingly large impact on prediction quality. Its innovation also forms one of the key ingredients of AlphaFold’s approach.
3 AlphaFold
Similar to RaptorX, AlphaFold predicts a distribution over discretized spatial ranges as its output (the details of the convolutional network architecture are different). Unlike RaptorX, which only exploits the mean and variance of the predicted distribution, AlphaFold uses the entire distribution as a (protein-specific) statistical potential function (Sippl, 1990; Thomas and Dill, 1996) that is directly minimized to fold the protein. The key idea of AlphaFold’s approach is that a distribution over pairwise distances between residues corresponds to a potential that can be minimized after being turned into a continuous function. DeepMind’s team initially experimented with more complex approaches (personal communication), including fragment assembly (Rohl et al., 2004) using a generative variational autoencoder (Kingma and Welling, 2013). Halfway through CASP13 however, the team discovered that simple and direct minimization of the predicted energy function, using gradient descent (L-BFGS) (Goodfellow et al., 2016; Nocedal, 1980), is sufficient to yield accurate structures.
There are important technical details. The potential is not used as is, but is normalized using a learned ‘reference state’. Human-derived reference states are a key component of knowledge-based potentials such as DFIRE (Zhang et al., 2005), but the use of a learned reference state is an innovation. This potential is coupled with traditional physics-based energy terms from Rosetta and the combined function is what is actually minimized. The idea of predicting a protein-specific energy potential is also not new (Zhao and Xu, 2012; Zhu et al., 2018), but AlphaFold’s implementation made it highly performant in the structure prediction context. This is important as protein-specific potentials are not widely used. Popular knowledge- and physics-based potentials are universal, in that they aspire to be applicable to all proteins, and in principle should yield a protein’s lowest energy conformation with sufficient sampling. AlphaFold’s protein-specific potentials on the other hand are entirely a consequence of a given protein’s MSA. AlphaFold effectively constructs a potential surface that is very smooth for a given protein family, and whose minimum closely matches that of the family’s average native fold.
Beyond the above conceptual innovations, AlphaFold uses more sophisticated neural networks than what has been applied in protein structure prediction. First, they are hundreds of layers deep, resulting in a much higher number of parameters than existing approaches (Liu et al., 2018; Wang et al., 2017). Second, through the use of so-called dilated convolutions, which use non-contiguous receptive fields that span a larger spatial extent than traditional convolutions, AlphaFold’s neural networks can model long-range interactions covering the entirety of the protein sequence. Third, AlphaFold uses sophisticated computational tricks to reduce the memory and compute requirements for processing long protein sequences, enabling the resulting networks to be trained for longer. While these ideas are not new in the deep learning field, they had not yet been applied to protein structure prediction. Combined with DeepMind’s expertise in searching a large hyperparameter space of neural network configurations, they likely contributed substantially to AlphaFold’s strong performance.
4 Future prospects
Much of the recent progress in protein structure prediction, including AlphaFold, has come from the incorporation of co-evolutionary data, which are by construction defined on the protein family level. For predicting the gross topology of a protein family, co-evolution-dependent approaches will likely show continued progress for the foreseeable future. However, such approaches are limited when it comes to predicting structures for individual protein sequences, such as a mutated or de novo designed protein, as they are dependent on large MSAs to identify co-variation in residues. Lacking a large constellation of homologous sequences, co-evolution-dependent methods perform poorly, and this was observed at CASP13 for some of the targets on which AlphaFold was tested (e.g. T0998). Physics-based approaches, such as Rosetta and I-Tasser, are currently the primary paradigm for tackling this broader class of problems. The advent of deep learning suggests a broader rethinking of how the protein structure problem could be tackled, however, with a broad range of possible new approaches, including end-to-end differentiable models (AlQuraishi, 2019; Ingraham et al., 2018), semi-supervised approaches (Alley et al., 2019; Bepler and Berger, 2018; Yang et al., 2018) and generative approaches (Anand et al., 2018). While not yet broadly competitive with the best co-evolution-dependent methods, such approaches can eschew co-evolutionary data to directly learn a mapping function from sequence to structure. As these approaches continue to mature, and as physico-chemical priors get more directly integrated into the deep learning machinery, we expect that they will provide a complementary path forward for tackling protein structure prediction.
Acknowledgements
We thank David Baker, Alex Bridgland, Jianlin Cheng, Richard Evans, Tim Green, John Jumper, Daisuke Kihara, John Moult, Sergey Ovchinnikov, Andrew Senior, Jinbo Xu and Augustin Zidekfor for lively discussions during CASP13 that formed the basis for much of the content of this letter. We also thank the anonymous reviewers for their constructive feedback which significantly improved the quality of our analysis.
Funding
This work was supported by NIGMS grant P50GM107618 and NCI grant U54-CA225088.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}