





Abstract
Protein–peptide interactions mediate a wide variety of cellular and biological functions. Methods for predicting these interactions have garnered a lot of interest over the past few years, as witnessed by the rapidly growing number of peptide-based therapeutic molecules currently in clinical trials. The size and flexibility of peptides has shown to be challenging for existing automated docking software programs.
Here we present AutoDock CrankPep or ADCP in short, a novel approach to dock flexible peptides into rigid receptors. ADCP folds a peptide in the potential field created by the protein to predict the protein–peptide complex. We show that it outperforms leading peptide docking methods on two protein–peptide datasets commonly used for benchmarking docking methods: LEADS-PEP and peptiDB, comprised of peptides with up to 15 amino acids in length. Beyond these datasets, ADCP reliably docked a set of protein–peptide complexes containing peptides ranging in lengths from 16 to 20 amino acids. The robust performance of ADCP on these longer peptides enables accurate modeling of peptide-mediated protein–protein interactions and interactions with disordered proteins.
ADCP is distributed under the LGPL 2.0 open source license and is available at http://adcp.scripps.edu. The source code is available at https://github.com/ccsb-scripps/ADCP.
Supplementary data are available at Bioinformatics online.
1 Introduction
Protein–peptide interactions are essential to many biological functions (Petsalaki and Russell, 2008). Thus, peptide-based therapeutic approaches have recently attracted increasing interest (Fosgerau and Hoffmann, 2015; Lau and Dunn, 2018). Moreover, many protein–protein interactions, especially the ones involving intrinsically disordered proteins, are mediated by peptide-like segments (Stein and Aloy, 2008; Wright and Dyson, 2015). Predicting protein–peptide interactions using automated docking methods remains challenging mainly due to the significantly larger number of rotatable bonds in peptides, making them more flexible than small drug-like molecules. Small molecule docking methods have been shown to perform rather poorly for peptides longer than five amino acids (Hauser and Windshügel, 2016; Rentzsch and Renard, 2015). Meanwhile, efforts have been put into developing accurate and efficient peptide docking methods (Ciemny et al., 2018; London et al., 2013). These methods can be segregated into the following three categories: template docking, ensemble docking and de novo methods (see Table 1).
Summary of three categories of peptide docking methods
| Category | Flexibility | Description | Examples |
|---|---|---|---|
| Template docking | None or little | Use sequence-based homology model to predict docking poses | GalaxyPepDock |
| Ensemble docking | Conformation ensemble | Prepare a conformation ensemble to describe peptide flexibility and then dock the conformations back into receptor |
|
| De novo method | Fully flexible | Model peptide flexibility with the respect to the receptor | ADCP
|
| Category | Flexibility | Description | Examples |
|---|---|---|---|
| Template docking | None or little | Use sequence-based homology model to predict docking poses | GalaxyPepDock |
| Ensemble docking | Conformation ensemble | Prepare a conformation ensemble to describe peptide flexibility and then dock the conformations back into receptor |
|
| De novo method | Fully flexible | Model peptide flexibility with the respect to the receptor | ADCP
|
Summary of three categories of peptide docking methods
| Category | Flexibility | Description | Examples |
|---|---|---|---|
| Template docking | None or little | Use sequence-based homology model to predict docking poses | GalaxyPepDock |
| Ensemble docking | Conformation ensemble | Prepare a conformation ensemble to describe peptide flexibility and then dock the conformations back into receptor |
|
| De novo method | Fully flexible | Model peptide flexibility with the respect to the receptor | ADCP
|
| Category | Flexibility | Description | Examples |
|---|---|---|---|
| Template docking | None or little | Use sequence-based homology model to predict docking poses | GalaxyPepDock |
| Ensemble docking | Conformation ensemble | Prepare a conformation ensemble to describe peptide flexibility and then dock the conformations back into receptor |
|
| De novo method | Fully flexible | Model peptide flexibility with the respect to the receptor | ADCP
|
The success of template docking methods for docking peptides (Lee et al., 2015) depends on the availability of homologue structures for both the receptor and the peptide, thus limiting the range of their applicability. Ensemble docking methods sample peptide conformations as a pre-processing step without knowledge of the receptor. Next, these conformations are docked rigidly or semi-rigidly into the receptors (Schindler et al., 2015; Yan et al., 2016; Zhou et al., 2018a, b). While, these methods yield good accuracy for small and medium sized peptides (typically ≤ 9 amino acids), their success rates tend to drop rapidly with longer peptides. Finally, de novo methods sample the peptide’s conformation on-the-fly during the docking (Alam et al., 2017; Ben-Shimon and Niv, 2015; Kurcinski et al., 2015; Raveh et al., 2011; Trellet et al., 2013). While de novo methods yield high accuracy and are less affected by the length of the peptides, these methods tend to be computationally expensive and often rely on lengthy molecular dynamics simulations to refine solutions.
Here we present AutoDock CrankPep or ADCP in short, an efficient de novo method for protein–peptide docking that folds the peptide in the potential energy landscape created by a given receptor. ADCP provides an efficient and accurate way to dock flexible peptides into rigid receptors. We show that it achieves 85.7% success rate on the LEADS-PEP dataset within its top 10 predictions. Furthermore, while existing peptide docking methods have typically limited themselves to peptides with less than 16 amino acids, we evaluate ADCP’s ability to dock a set of peptides ranging in length from 16 to 20 amino acids. For these peptides, ADCP achieves re-docking success rates of 64% and 91% when considering the top or top 5 solutions, respectively. These results indicate that ADCP is a robust peptide docking tool that can be used to model protein–protein interactions mediated by protein segments such as loops or disordered fragments.
2 Materials and methods
Small molecule docking methods typically perform best with ligands containing less than 20 rotatable bonds (Hauser and Windshügel, 2016; Rentzsch and Renard, 2015). Peptides with five or more amino acids can easily exceed this number. A medium sized peptide of 10 amino acids typically has around 40 rotatable bonds, rendering these methods ineffective.
CRANKITE is an efficient software package originally developed for protein and peptide conformation sampling and folding (Burkoff et al., 2012; Podtelezhnikov and Wild, 2005, 2008; Várnai et al., 2013). It samples the conformational space of proteins or peptides using a Metropolis Monte Carlo (MC) search and a Gō-Type representation of amino acid side-chains (Takada, 1999; Taketomi et al., 2009). CRANKITE can rapidly explore the conformational space of sequences of amino acids by performing the two MC moves illustrated in Figure 1: (i) a crankshaft motion along two randomly selected Cα atoms and (ii) a rotation near the end of the chain.
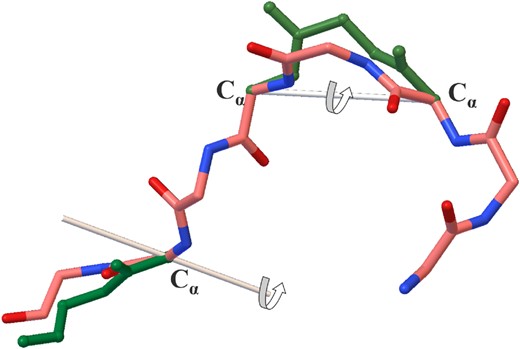
CRANKITE’s Monte Carlo moves. A crankshaft motion along two selected Cα atoms or a rotation near the end of the chain
ADCP combines CRANKITE’s conformation sampling ability with the grid-based AutoDock representation of a rigid receptor (Huey et al., 2007; Morris et al., 2009) to concurrently optimize the peptide conformation and its interactions with the receptor, thus yielding docking poses. ADCP was implemented based on CRANKITE. The notable modifications and additions are as follows: (i) the addition of new MC moves to boost the exploration of peptide position and orientation relative to the receptor; (ii) the addition of an energy term based on the AutoDock affinity grids to describe the peptide-receptor interactions; (iii) the use of a rotamer library (Dunbrack Jr and Cohen, 1997) to interactively construct side-chain atoms; and (iv) the addition of a pose cache swapping mechanism to enhance the search.
The overall workflow of the MC procedure implemented by ADCP is depicted in Figure 2. First, a randomly selected MC move is applied to alter the current pose. The altered pose is then scored, and the move is either accepted or rejected based on a metropolis-like MC criterion. If the move is rejected, the pose before the move is restored and another move is attempted. If it is accepted, the altered pose becomes the current one and is used to update the cache of docking poses. This procedure repeats until one of the termination criteria is met. More details about the various elements of this workflow are provided below.
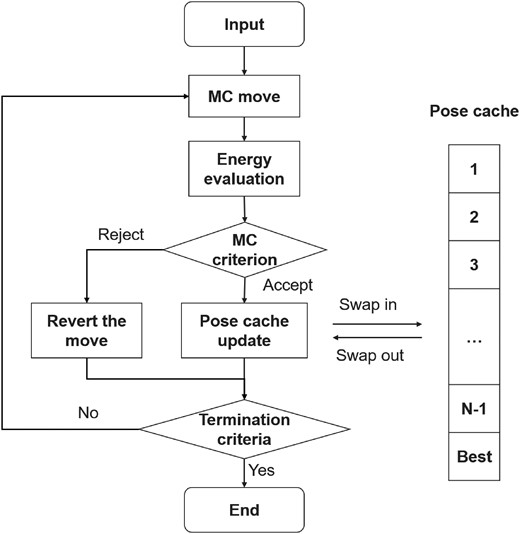
Flow chart of AutoDock CrankPep Monte Carlo procedure
Input : ADCP requires a description of the receptor and the peptide. The receptor is represented by affinity maps calculated using AutoGridFR (Ravindranath et al., 2015). AutoGridFR produces a single zip file that contains affinity maps for all atom types in the peptide calculated by AutoGrid4 (Morris et al., 2009), along with metadata about the docking box (e.g. the size and position of the box, a list of favorable locations in the affinity maps called translational points, etc.). The peptide can be specified as a 3D structure in the PDB file format or by its sequence of amino acids. In the latter case, a starting conformation is constructed automatically. This initial conformation can be generated in an extended or alpha helical conformation using lowercase and uppercase letters respectively. The user can also specify the maximum number of MC steps for the simulation. As conventional docking methods can dock short peptides with reasonable accuracy, ADCP was designed to support peptides with five or more amino acids.
Monte Carlo moves : During the MC search the docking pose is modified using MC moves. We extended the original set of MC moves with: (i) a local translation to perturb the peptide position; and (ii) a translational jump that translates the peptide to move a central peptide atom called the ‘root atom’ to a new ‘translational point’ in the docking box (Supplementary Fig. S1). The ‘translational points’ are a set of positions with high affinities identified by AutoGridFR (Ravindranath and Sanner, 2016). These moves were added to facilitate the peptide’s exploration of the potential field created by the receptor. Each MC move will trigger the reconstruction of the all-atom representation of the side-chains using a rotamer library (see below).
Scoring Function : The scoring function of ADCP consists of two components: a score for the conformation of the peptide; and a score for the interaction between the peptide and the receptor. We will refer to these scores as the internal and the interaction scores, respectively. The internal score is based on CRANKITE’s Gō-Type potential (Podtelezhnikov and Wild, 2005, 2008) where each side-chain is represented by a single bead. We extended the internal score function with a new term based on Ramachandran propensities for backbone φ and ψ angles (Lovell et al., 2003). These Ramachandran propensities are transformed into energies according to the Boltzman distribution. The interaction score between the peptide and the protein is calculated using the AutoDock affinity grids. Calculating this score requires a full-atom representation of the peptide which is constructed using a rotamer library (Dunbrack Jr. and Cohen, 1997). Every time a pose is scored, we iterate over the peptide either forward (N-to C-terminus) or backwards (C- to N-terminus) with the same probability. For each amino acid, we construct all rotameric conformations and score them using the affinity grids while avoiding clashes with the peptide backbone and already built side-chains. The energetically most favorable side-chain conformation is selected to represent this amino acid. Once the full-atom representation of the peptide is built, the interaction score between the peptide and the receptor is obtained by summing up the scores from the affinity grids for all atoms in the peptide.
Pose cache update : The presence of the receptor creates a complex energy landscape for the peptide to fold while maximizing its interaction with the receptor. We found that the traditional MC search often got stuck in local minima. Thus, we implemented the pose cache to maintain a pool of docking poses encountered during the search and restart the search under certain circumstances. Every pose accepted by the Metropolis criterion is compared with the ones present in the cache and can: either be added to the cache (e.g. if it has the best energy found so far) or it can replace an entry in the cache (e.g. a similar solution is in the cache but with a worse energy). More details on this process are provided in Chart S1. Every time a pose improves on the best score seen so far, it is appended to the output multi-model pdb file. If the search generates 100 000 consecutive poses each having a score 3 kcal/mol higher than the best score found so far, or the best score has not improved for one million steps, the search restarts from a randomly selected pose from the cache.
Termination criteria: The program stops if the maximum number of steps is reached or the best score has not been improved for 10 million steps.
Using the software: The affinity maps are calculated using AutoGridFR for a user-defined docking box. The results presented here were docked with a 4 Å padding on every side of the peptide. We also performed redockings with 6 and 8 Å padding, the latter often covering the bulk of the receptor. While we observed minor changes in the ordering of the docking poses due to the stochastic nature of the algorithm, these changes do not modify the reported sampling success rate. The stochastic nature of the MC search is usually addressed by performing multiple searches called replicas. The number of replicas is specified by the user in ADCP. Unless otherwise specified, the results presented here were obtained using 80 replicas with 60 simulations started from extended conformations and 20 simulations started from helical conformations constructed from amino acid sequence. Each replica performed 3 million MC steps per amino acid in the peptide (i.e. 15 million steps for a 5-mer peptide and 36 million steps for 12-mer peptide, etc.). While we routinely observed conformational changes between helical conformation and extended conformation and vice versa during the MC runs, we found that statistically, starting the MC with a mix of initial conformation speeds up the process of identifying the correct solution. Users can customize these parameters according to their specific needs. We suggest using more replicas and longer simulations for larger peptides and/or larger docking boxes.
After all replicas finish their search, the docking poses are clustered to produce the final docking poses. The clustering can be performed using the AutoDockFR clustering algorithm (Morris et al., 2009; Ravindranath et al., 2015), or using pairs of peptide-receptor residues in contact. See the Supplementary Information for a detailed description of the contact-based clustering algorithm.
2.1 Datasets
The peptiDB (London et al., 2010) and LEADS-PEP (Hauser and Windshügel, 2016) protein–peptide datasets have been widely used for benchmarking peptide docking methods (Raveh et al., 2011; Tubert-Brohman et al., 2013; Zhou et al., 2018a). PeptiDB contains 102 protein–peptide complexes varying from 3 to 15 amino acids. We benchmarked ADCP with the Glide SP-PEP dataset (Tubert-Brohman et al., 2013), a subset of peptiDB comprised of 19 high-quality, non-α-helical complexes ranging from 5 to 11 amino acids. The Glide SP-PEP dataset has been used to benchmark FlexPepDock, Glide SP-PEP and HPepDock. The LEADS-PEP dataset is a more recent, and manually curated dataset of 53 complexes with peptides ranging from 3 to 12 amino acids. In this study we used the subset of 42 complexes from LEADS-PEP containing peptides with 5 or more amino acids. We consider the peptides in these datasets as medium-sized peptides for docking purposes.
Current available peptide docking methods are mostly tested on peptides with 15 amino acids or less. To further test ADCP, we compiled at set of 11 protein peptide complexes from the protein data bank (PDB) (Berman et al., 2000) with longer peptides ranging in length from 16 to 20 amino acids. These structures were obtained by selecting PDB entries with crystallographic resolution of 2.2 Å or better and containing a peptide comprised of 16–20 standard amino acids. The peptides in this set are neither cyclic nor covalently bound to the receptor; they have no significant clashes between peptide and receptor atoms and have no significant contacts between the peptide and crystal mates of the receptor. These complexes are listed in Supplementary Table S1.
We found apo structures for 5 of the 11 protein–peptide complexes (Supplementary Table S1) by retrieving all PDB entries sharing the uniport accession code of the protein in the protein–peptide complex (holo protein). The sequences of the apo-candidates and holo proteins were then aligned to the UNIPROT (UniProt Consortium, 2019) sequence yielding a mapping of the apo-candidate sequence to the holo sequence. We only retained entries with 70% or more of the apo sequence mapped to the holo sequence. Next, we structurally aligned the apo-candidate structure with the holo protein and we identified overlaps between the peptide and atoms in the apo-candidate structure. We retained entries in which the peptide only overlap with water, ions , additives or apo receptor chain. For the three complexes 2IVZ, 5UWI and 6CIT, which resulted in more than one apo structure (2, 12 and 12 respectively), we selected the apo structure with the best crystallographic resolution.
2.2 Success metrics
All atom Root-Mean-Square Deviation (RMSD) is typically used to assess success while docking small molecules. As ligands grow larger this metric becomes less appropriate and RMSD of backbone atoms (N, CA, C) has been used for assessing docking success rate for small peptides (Irving et al., 2001; Méndez et al., 2003). For instance, (Hauser and Windshügel, 2016; Zhou et al., 2018a) used a 2.5 Å backbone RMSD cutoff to define successful peptide redocking on the Lead-Pep dataset. Other studies (Raveh et al., 2011; Tubert-Brohman et al., 2013) used the iRMSD (interface RMSD) defined as the RMSD of the backbone atoms of the ‘interface residues’. Interface residues are amino acids of the peptide having their Cβ atom within 8 Å of any receptor Cβ atom. Poses with iRMSD values under 3.0 Å are typically considered to be successful dockings. To facilitate the comparison with other methods, we used the same metric as used in previously published studies, i.e. backbone RMSD for comparison on the LEADS-PEP dataset and iRMSD for the Glide SP-PEP dataset.
When docking longer peptides, RMSD-based metrics do not provide a precise measure for success. For these cases, we assess success using native contacts: a metric borrowed from the protein–protein docking field (Irving et al., 2001; Méndez et al., 2003). Native contacts are defined as the list of all pairs of peptide-receptor amino acids located within 5 Å of each other. Similar to (Méndez et al., 2003; Peterson et al., 2017; Yan et al., 2016), we identify successful redockings of peptides ranging from 16 to 20 amino acids in length when the docking poses reproduce more than 50% of the native contacts (i.e. fnc > 0.5).
3 Results and discussion
We compared the success rate of ADCP with previously published results from leading peptide docking methods on the datasets containing medium size peptides (i.e. up to 12 amino acids). We also demonstrate that ADCP achieves remarkable success rates in docking longer peptide (i.e. up to 20 amino acids).
3.1 Accurate docking of medium size peptides
HPepDock (Zhou et al., 2018a, b) is a recent peptide docking method that falls into the ensemble docking category. It uses MODPEP (Yan et al., 2017) to generate 1000 peptide conformations and then docks these peptide conformations semi-rigidly using MDock (Huang and Zou, 2006). HPepDock has been shown to achieve better accuracy than traditional small-molecule docking methods as well as other leading peptide docking methods including FlexPepDock, Glide-SP-PEP, HADDOCK and pepATTRACT (Zhou et al., 2018a, b). As such, it can be considered the state of the art at the time of writing this paper. Here we compare the success rates of ADCP with the HPepDock results on these datasets using the same metrics. For the complexes from the LEADS-PEP dataset, ADCP consistently outperforms HPepDock as shown in Figure 3. Considering the top 10 solutions, ADCP achieves 85.7% success rate for this dataset compared to 66.7% for HPepDock at 2.5 Å RMSD cutoff. ADCP significantly improves success rate at 1.0 Å RMSD cutoff, predicting 76.2% of the complexes with a sub-angstrom backbone RMSD precision within its top 1000 predictions, and 100% of the complexes have a correctly docked pose (backbone RMSD ≤ 2.5 Å). The complex-specific comparison is provided in Supplementary Table S2.
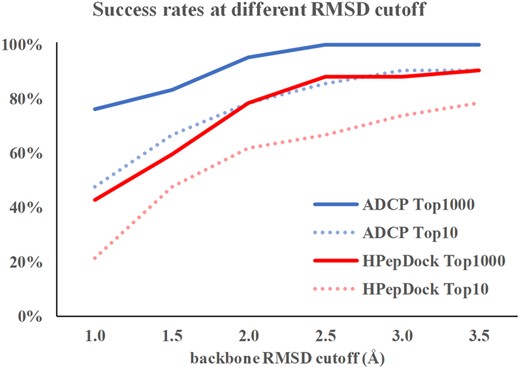
The comparison of success rates for ADCP and HPepDock with different success criteria. Blue solid line represents the success rate for ADCP if the top 1000 solutions are considered and blue dashed line represents the success rate if the top 10 solutions are considered. Red solid and dashed lines represent the success rate for HPepDock if the top 1000 solutions and the top 10 solutions are considered, respectively (Color version of this figure is available at Bioinformatics online.)
HPepDock’s performance deteriorates significantly for the longer peptides in this dataset (Table 2). While it’s overall success rate at 2.5 Å backbone RMSD for the top 10 solution is 66.7% (28/42), when considering only peptides with 9 or more amino acids the success rate drops to 35% (7/20). ADCP on the other hand maintains its 85% overall success rate on the subset of peptides with 9 or more amino acids.
Success rates at 2.5 Å backbone RMSD considering the top 10 solutions
| bbRMSD ≤ 2.5 Å | ADCP | HPepDock |
|---|---|---|
| All | 85.7% | 66.7% |
| Longer peptides | 85.0% | 35.0% |
| bbRMSD ≤ 2.5 Å | ADCP | HPepDock |
|---|---|---|
| All | 85.7% | 66.7% |
| Longer peptides | 85.0% | 35.0% |
Note: Out of the 42 peptides, 20 have more than 8 amino acids (48%) and are classified as ‘longer peptides’.
Success rates at 2.5 Å backbone RMSD considering the top 10 solutions
| bbRMSD ≤ 2.5 Å | ADCP | HPepDock |
|---|---|---|
| All | 85.7% | 66.7% |
| Longer peptides | 85.0% | 35.0% |
| bbRMSD ≤ 2.5 Å | ADCP | HPepDock |
|---|---|---|
| All | 85.7% | 66.7% |
| Longer peptides | 85.0% | 35.0% |
Note: Out of the 42 peptides, 20 have more than 8 amino acids (48%) and are classified as ‘longer peptides’.
A possible explanation for this could be that for longer peptides, the conformational space, which increases exponentially with the length of the peptide, eventually requires a prohibitively large numbers of conformers to be used in ensemble docking methods. ADCP explores the peptide's conformational space during the docking process thus it is less affected by increasing peptide lengths. ADCP maintains a consistent success rate across the peptides lengths in this dataset.
On the smaller Glide SP dataset, ADCP performs similarly to other methods for both holo (19 complexes) and apo (10 complexes) receptor conformations, as shown in Table 3.
Success rates on the Glide SP-PEP dataset
| iRMSD | ADCP | FlexPepDock | Glide SP-PEP | HPepDock | |
|---|---|---|---|---|---|
| holo | ≤2 Å | 13 | 13 | 11 | 12 |
| ≤3 Å | 15 | 13 | 13 | 15 | |
| apo | ≤2 Å | 4 | 6 | 4 | 5 |
| ≤3 Å | 8 | 6 | 4 | 8 | |
| iRMSD | ADCP | FlexPepDock | Glide SP-PEP | HPepDock | |
|---|---|---|---|---|---|
| holo | ≤2 Å | 13 | 13 | 11 | 12 |
| ≤3 Å | 15 | 13 | 13 | 15 | |
| apo | ≤2 Å | 4 | 6 | 4 | 5 |
| ≤3 Å | 8 | 6 | 4 | 8 | |
Note: Among the 19 complexes in the dataset, 10 receptors have apo conformation available. Here a docking is deemed successful if one of the top 10 solutions has an interface RMSD (iRMSD) lower than 2.0 Å or 3.0 Å respectively.
Success rates on the Glide SP-PEP dataset
| iRMSD | ADCP | FlexPepDock | Glide SP-PEP | HPepDock | |
|---|---|---|---|---|---|
| holo | ≤2 Å | 13 | 13 | 11 | 12 |
| ≤3 Å | 15 | 13 | 13 | 15 | |
| apo | ≤2 Å | 4 | 6 | 4 | 5 |
| ≤3 Å | 8 | 6 | 4 | 8 | |
| iRMSD | ADCP | FlexPepDock | Glide SP-PEP | HPepDock | |
|---|---|---|---|---|---|
| holo | ≤2 Å | 13 | 13 | 11 | 12 |
| ≤3 Å | 15 | 13 | 13 | 15 | |
| apo | ≤2 Å | 4 | 6 | 4 | 5 |
| ≤3 Å | 8 | 6 | 4 | 8 | |
Note: Among the 19 complexes in the dataset, 10 receptors have apo conformation available. Here a docking is deemed successful if one of the top 10 solutions has an interface RMSD (iRMSD) lower than 2.0 Å or 3.0 Å respectively.
3.2 Reliable docking long peptides and protein segments
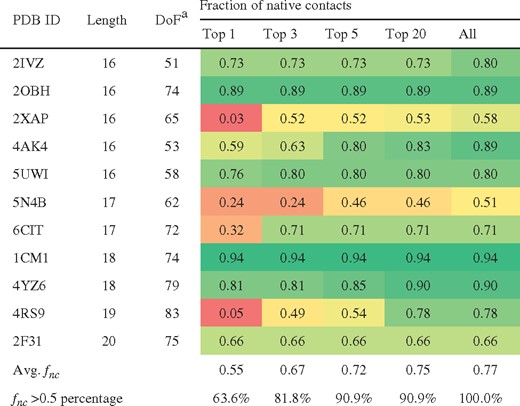
Currently available peptide docking methods have mostly been tested and benchmarked on small and medium-sized peptides with up to 15 amino acids in length. However, a considerable portion of protein–protein interactions are mediated by flexible protein loops, disordered chains segments, or intrinsically disordered proteins, involving sequences that can easily exceed 15 amino acids. Therefore, we tested ADCP on a set of 11 complexes containing peptides with 16–20 amino acids in length. For these dockings we performed 80 MC simulations, allotting 7×N million MC moves (where N is the number of amino acids in the peptide) to each run. The docking poses from the 80 MC runs were clustered using contacts with a cutoff value of 80%. Results are shown in Table 4.
Docking results for long peptides and protein segments
 |
|
DoF: degrees of freedom including translation, rotation and rotatable bonds.
The cells are colored based on fnc from red (0.0) to yellow (0.5) and to green (1.0).
Docking results for long peptides and protein segments
|
|
DoF: degrees of freedom including translation, rotation and rotatable bonds.
The cells are colored based on fnc from red (0.0) to yellow (0.5) and to green (1.0).
Considering only the top-ranking solution, ADCP identifies solution with at least 50% native contacts for 7 out of 11 complexes (63.6%). Within the top 5 solutions, the success rates increase to 90.9% (10 out of 11 complexes). Figure 4 shows some examples of docked pose with respect to the crystal structure along with the fraction of reproduced native and backbone RMSD values.
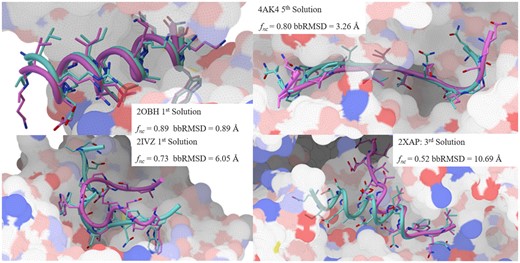
Comparison of selected docked poses (cyan) and experimentally determined structures (magenta). The receptor surface is provided for context and colored using a polarity scheme developed by Dr. D. Goodsell. These Figures are generated using PMV 1.5.7 (Sanner, 1999) and MSMS (Sanner et al., 1996) (Color version of this figure is available at Bioinformatics online.)
For the 5 complexes for which apo structures are available, ADCP obtained good solutions for 2 complexes with fnc of more than 0.5 and partially docked solutions for 2 complexes with fnc of more than 0.3 within its 5 predictions. Docking failed for the 1CM1 complex in which the protein forms a tunnel where a substantial conformational change in the apo structure greatly shrinks the width of the tunnel, preventing the peptide from binding there. See Supplementary Table S3 for more details.
ADCP demonstrates a robust ability to dock long peptides and protein segments. With more MC replicas and more steps for each replica, ADCP could potentially be applied to even longer peptide-like segments.
It is noteworthy that the current scoring function in ADCP has not been optimized or calibrated for protein–peptide interactions. The interaction energy between peptide and receptor relies on AutoDock4 affinity maps that were initially developed and calibrated for docking small, drug-like molecules. While we have started incorporating peptide-specific elements, such as a potential for Ramachandran backbone angles into our scoring function, further refinements of the current scoring function could improve docking success rates. Alternatively, re-scoring top-ranked poses with scoring functions designed for protein–peptide interactions (Huang and Zou, 2008; Spiliotopoulos et al., 2016) could also improve the ranking of the docking predictions.
3.3 Timing
ADCP is computationally efficient compared with other de novo methods. Based on 3 million MC moves per amino acid, a MC search typically takes from ∼10 min for a 5-mer to ∼1 h for a 12-mer, using a single thread on an Intel Xeon E5-1620 processor (2014). These times are rough averages and can vary depending on the peptide sequence. See Supplementary Table S2 for detailed timing results on LEADS-PEP dataset. Each MC simulation can be run independently and they can execute in parallel locally or on high performance computing clusters. The time for the final clustering is a function of the clustering cutoff but typically only takes a few minutes. While HPepDock and other ensemble docking methods requires less computational resources, ADCP achieves better success rates especially for longer peptides. HPepDock remains a powerful peptide docking tool for medium size peptides.
4 Conclusions
In this paper, we presented ADCP, a novel approach for predicting protein–peptide interactions for peptides of substantial length. The approach leverages an algorithm developed initially for protein folding and combines it with a representation of a rigid receptor developed for docking. With a success rate of 85.7% on the LEADS-PEP dataset when considering the top 10 predictions, ADCP outperforms leading peptide docking approaches. Moreover, we show that ADCP is able to dock peptides with up to 20 amino acids to their receptors. ADCP expands peptide docking to the prediction of certain types of protein–protein interactions, e.g. disordered tails or flexible protein loops interacting with itself or another protein.
Acknowledgements
The authors thank Drs Wild, Podtelezhnikov and Várnai for the original implementation of CRANKITE and many helpful discussions and suggestions. The authors also thank the members of the Center for Computational Structural Biology for many fruitful discussions. This is manuscript 29784 from The Scripps Research Institute.
Funding
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM096888 to Dr. M. F. Sanner. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
References
UniProt Consortium (


{kind=link}
{kind=link}
{kind=link}
{kind=link}