





Abstract
The vast majority of tools for neoepitope prediction from DNA sequencing of complementary tumor and normal patient samples do not consider germline context or the potential for the co-occurrence of two or more somatic variants on the same mRNA transcript. Without consideration of these phenomena, existing approaches are likely to produce both false-positive and false-negative results, resulting in an inaccurate and incomplete picture of the cancer neoepitope landscape. We developed neoepiscope chiefly to address this issue for single nucleotide variants (SNVs) and insertions/deletions (indels).
Herein, we illustrate how germline and somatic variant phasing affects neoepitope prediction across multiple datasets. We estimate that up to ∼5% of neoepitopes arising from SNVs and indels may require variant phasing for their accurate assessment. neoepiscope is performant, flexible and supports several major histocompatibility complex binding affinity prediction tools.
neoepiscope is available on GitHub at https://github.com/pdxgx/neoepiscope under the MIT license. Scripts for reproducing results described in the text are available at https://github.com/pdxgx/neoepiscope-paper under the MIT license. Additional data from this study, including summaries of variant phasing incidence and benchmarking wallclock times, are available in Supplementary Files 1, 2 and 3. Supplementary File 1 contains Supplementary Table 1, Supplementary Figures 1 and 2, and descriptions of Supplementary Tables 2–8. Supplementary File 2 contains Supplementary Tables 2–6 and 8. Supplementary File 3 contains Supplementary Table 7. Raw sequencing data used for the analyses in this manuscript are available from the Sequence Read Archive under accessions PRJNA278450, PRJNA312948, PRJNA307199, PRJNA343789, PRJNA357321, PRJNA293912, PRJNA369259, PRJNA305077, PRJNA306070, PRJNA82745 and PRJNA324705; from the European Genome-phenome Archive under accessions EGAD00001004352 and EGAD00001002731; and by direct request to the authors.
Supplementary data are available at Bioinformatics online.
1 Introduction
While mutations may promote oncogenesis, cancer-specific variants and the corresponding novel peptides they may produce (‘neoepitopes’) appear central to the generation of adaptive anti-tumor immune response (Efremova et al., 2017). The advent of immunotherapy as a promising form of cancer treatment has been accompanied by a parallel effort to explore tumor neoepitopes as potential mechanisms and drivers of therapeutic response. This has prompted the development of numerous neoepitope prediction pipelines. Initial bioinformatics approaches used tumor-specific sequencing data to focus on missense single nucleotide variants (SNVs) as the predominant source of neoepitopes [e.g. Epi-Seq (Duan et al., 2014)]. However, as ∼15% of neoepitopes is estimated to result from other types of mutations (Vogelstein et al., 2013), additional tools were developed to predict neoepitopes from gene fusions [e.g. INTEGRATE-neo (Zhang et al., 2017)], non-stop mutations [e.g. TSNAD (Zhou et al., 2017)], and insertions and deletions [indels, from e.g. pVACseq (Hundal et al., 2016), MuPeXI (Bjerregaard et al., 2017)], which may be of particular significance for anticipating cancer immunotherapy response (Turajlic et al., 2017). Many of these tools enable comparable predictive capabilities, but each approach has its own unique set of features and limitations (see Figure 1).
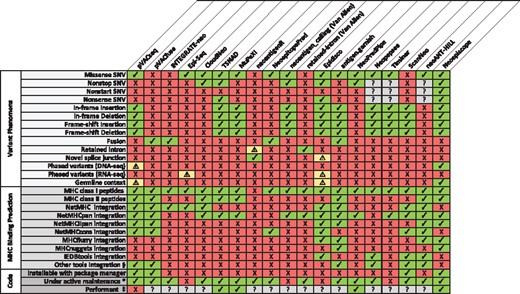
A feature comparison of pVACseq and pVACfuse (Hundal et al., 2016), INTEGRATE-neo (Zhang et al., 2017), Epi-Seq (Duan et al., 2014), CloudNeo (Bais et al., 2017), TSNAD (Zhou et al., 2017), MuPeXI (Bjerregaard et al., 2017), neoantigenR (Tang and Madhavan, 2017), NeoepitopePred (Chang et al., 2017), neoantigen_calling_pipeline (Van Allen laboratory; (Van Allen et al., 2015), retained-intron-neoantigen-pipeline (Van Allen laboratory; Smart et al., 2018), Epidisco (Rubinsteyn et al., 2017), antigen.garnish (Rech et al., 2018), NeoPredPipe (Schenck et al., 2018), Neopepsee (Kim et al., 2018), TIminer (Tappeiner et al., 2017), ScanNeo (Wang et al., 2019), neoANT-HILL (Coelho et al., 2019) and neoepiscope. Each column corresponds to a software tool, with software features listed by row. A checkmark indicates that the tool possesses or processes the indicated feature, whereas a ‘X’ indicates that the tool does not possess or process the indicated feature. A warning symbol (‘!’) indicates that a tool incompletely supports the corresponding feature. Specifically, neoantigenR can only identify retained introns when using GTF annotations that contain labeled introns. Epidisco is limited to RNA-seq data and cannot account for downstream effects of a variant, though it can identify some events of alternative splicing, germline variation and nearby somatic variation so long as they occur nearby a declared variant. Epi-Seq uses phasing information to determine the genotypes of variants and to identify other variants nearby variants that produce neoepitopes, but like Epidisco it does not take into account the downstream effects of a variant. pVACseq is able to perform variant phasing for substitutions including proximal germline variants using GATK ReadBackedPhasing (Hundal et al., 2019). Question marks (‘?’) denote unknown or unassessed values. *A tool was considered to be under activate maintenance if a new release or GitHub commit had occurred within 6 months prior to submission of this manuscript. §Other MHC binding prediction tools used include NNalign, PickPocket, SMM, SMMPMBEC and SMM align for pVACseq and pVACfuse; NetMHCII for antigen.garnish, and NetCTLpan for Neopepsee. ‡A tool was considered to be performant if neoepitope prediction averaged <10 min per sample in our benchmarking (see Materials and Methods section)
However, a limitation of most neoepitope prediction tools is that they consider individual somatic variants in the sequence context of a single reference genome, neglecting interactions between somatic and potentially neighboring germline variants given the widespread genetic variability among individuals (1000 Genomes Project Consortium et al., 2010). Indeed, somatic and germline mutations may co-occur even within the three nucleotide span of a single codon (Koire et al., 2017). Such immediate co-occurrences are not uncommon, affecting 17% of cancer patients on average, and can significantly alter variant effects including the amino acid sequence of any predicted neoepitope. Somatic mutations may also cluster or co-occur within a short span of each other. For instance, several hundred somatic mutations were found to co-occur within a <10 bp intervariant distance among whole-genome sequencing data from two breast cancer patients (Muiño et al., 2014). Furthermore, immediate co-occurrences of variants are not a prerequisite for phasing to be relevant to neoepitope prediction. For example, frameshifting indels upstream of a SNV can alter the peptide sequence in the vicinity of the SNV, as well as the consequences of the SNV itself.
The vast majority of neoepitope prediction tools only consider single somatic variants in isolation from each other, neglecting the potential for these compound effects. We developed neoepiscope chiefly to incorporate germline context and address variant phasing for SNVs and indels, and herein, illustrate how these features affect neoepitope prediction across multiple patient datasets.
2 Materials and Methods
2.1 Software
2.1.1 Preprocessing
The neoepiscope neoepitope prediction pipeline is depicted in Figure 2. It is highly flexible and intentionally agnostic to the user’s preferred tools for alignment, human leukocyte antigen (HLA) typing, variant calling and major histocompatibility complex (MHC) binding affinity prediction. The pipeline upstream of neoepiscope takes as input DNA-seq (exome or whole genome) FASTQs for a tumor sample and a matched normal sample. These FASTQs are used to perform HLA genotyping, e.g. using OptiType (Szolek et al., 2014). The FASTQs are also aligned to a reference genome with a DNA-seq aligner such as BWA (Li and Durbin, 2010) or Bowtie 2 (Langmead and Salzberg, 2012). The resulting alignments are subsequently used to call somatic and, optionally, germline variants. Somatic variants are obtained by running any somatic variant caller or combination of callers on the tumor and normal sample’s alignments, acknowledging that consensus-based approaches give more accurate results than a single caller alone (Ewing et al., 2015). To obtain most of our results, we used a combination of Pindel, MuSE, MuTect, RADIA, SomaticSniper and VarScan 2 (see Section 2.2). Germline variants are obtained by running a germline variant caller such as Genome Analysis Toolkit’s (GATK) HaplotypeCaller (McKenna et al., 2010) or VarScan 2 (Koboldt et al., 2012) on the normal sample’s alignments. Next, somatic variants are phased, optionally with germline variants, by a variant phasing tool such as HapCUT2 (Edge et al., 2017; note that neoepiscope was designed to not only be run with HapCUT2-formatted input, but can also be run with a phased Variant Call Format file (VCF) produced by GATK’s ReadBackedPhasing or an unphased VCF). The resulting phased variants (‘phase sets’) are input to neoepiscope together with a gene transfer format (GTF) file encoding known transcript variants (e.g. GENCODE). neoepiscope can be run with variants predicted from a variety of reference genome builds—the software has built-in functionality for handling GENCODE’s GRCh37 (v19) and GRCh38 (v29) human reference genome builds, but allows for users to input variants from any reference genome of their choice (human or otherwise), so long as they provide a GTF file and Bowtie index (Langmead et al., 2009) for that genome.
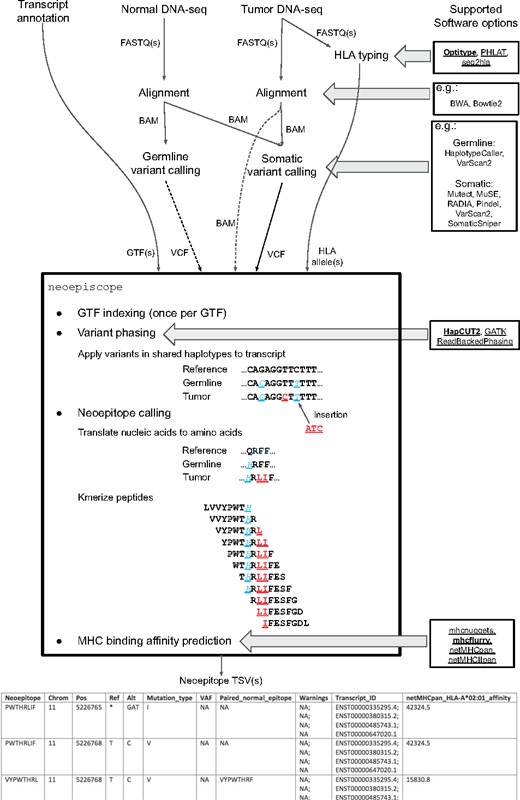
Neoepitope prediction pipeline diagram describing canonical neoepiscope workflow. Global inputs are shown at the top of the figure, with connecting arrows demonstrating interim inputs and outputs between preprocessing and processing steps. Direct inputs to and outputs from neoepiscope are shown directly entering or leaving the outlined box listing neoepiscope functionality. Multiple potential software options are shown at right for each relevant processing step as indicated by horizontal arrows (tools that are directly compatible with neoepiscope are underlined, with those in bold implemented as default). Direct neoepiscope functionality is depicted within the outlined box, with example sequences showing both somatic (underlined) and background germline variants (underlined, italic) in a mock transcript sequence, and their translation and kmerization into short peptides (8-mers). An example of the resulting neoepiscope output is shown at the bottom
2.1.2 Neoepitope calling
The principal contribution of this manuscript is the neoepitope calling component of neoepiscope. Before it is run on the phase sets associated with a given tumor–normal sample pair, neoepiscope indexes the GTF into a set of pickled dictionaries that link exon, start codon and stop codon blocks from transcripts to the genomic intervals they cover. These dictionaries can be reused with every call to neoepiscope to reduce runtime. Prior to neoepitope prediction, unphased somatic variants from the somatic VCF are added to HapCUT2 output as their own haplotype blocks using neoepiscope’s prep mode. For each haplotype block, neoepiscope determines if any mutation in the block overlaps any transcript(s) in the annotation, and then applies each mutation to the transcript. [Transcript sequences are obtained by concatenating relevant genomic sequence from an appropriate Bowtie (Langmead et al., 2009) index.] By default, all relevant somatic and germline variants in a haplotype block are applied, but users may choose to exclude either germline or somatic variants or to exclude variant phasing entirely. With all variants applied together, the synergistic effects of variants, such as those affecting the same codon, can be relayed. Each transcript is translated in silico, proceeding from the annotated start codon (or the first annotated codon if no start is present), but this process may be modified via command-line options to perform alternative start codon predictions. The resulting protein is kmerized in the vicinity of amino acid changes, and epitopes that do not match any peptide sequence in the normal protein are returned as potential neoepitopes. We performed extensive unit testing on our software to ensure that both common and rare cases of somatic variants are handled accurately. Patient-specific HLA alleles and a choice of MHC binding prediction tools can be specified so that optional binding prediction can be performed for each potential neoepitope. As indicated in Figure 2, neoepiscope itself calls the user-specified MHC binding affinity prediction software to obtain HLA allele-associated binding affinities. By default, it installs MHCflurry (O’Donnell et al., 2018) and MHCnuggets (Bhattacharya et al., 2017). The user may also independently install NetMHCpan (O’Donnell et al., 2018) or NetMHCIIpan (Nielsen et al., 2008) for use with neoepiscope. The output of neoepiscope is a TSV file listing one neoepitope per line in alphabetical order. For each neoepitope, information is given about the mutation of origin, the VAF (if available), the paired normal epitope for SNV- or MNV-derived epitopes, transcript identifiers for source transcripts (as well as warnings associated with those transcripts if relevant, e.g. disruptions to the reference start codon), and binding affinity information for all alleles and prediction tools specified. An example of the output is available in the neoepiscope GitHub repository (https://github.com/pdxgx/neoepiscope).
2.2 Variant identification and phasing
We assembled a cohort of 375 tumor samples from 347 different cancer patients from publicly available data including 285 melanoma patients (313 tumor samples; Amaria et al., 2018; Carreno et al., 2015; Gao et al., 2016; Hugo et al., 2017; Roh et al., 2017; Snyder et al., 2014; Van Allen et al., 2015; Zaretsky et al., 2016), 34 non-small cell lung cancer patients (28 tumor samples; Rizvi et al., 2015), and 28 colon, endometrial and thyroid cancer patients (29 tumor samples; Le et al., 2017; see Supplementary Table S1). Whole-exome sequencing (WES) reads for each sample were aligned to GRCh37d5 using the Sanger cgpmap workflow (cancerit, n.d.), which uses bwa-mem (v0.7.15-1140) (Li and Durbin, 2009) and biobambam2 (v2.0.69; gt, n.d.) to generate genome-coordinate sorted alignments with duplicates marked and GATK (v3.6) to realign around indels and perform base recalibration for paired tumor and normal sequence read data. Somatic variants were called and filtered using MuSE (v0.9.9.5; Fan et al., 2016), MuTect (v1.1.5; Cibulskis et al., 2013), Pindel (v0.2.5b8; Ye et al., 2009), RADIA (v1.1.5; Radenbaugh et al., 2014), SomaticSniper (v1.0.5.0; Larson et al., 2012) and VarScan 2 (v2.3.9; Koboldt et al., 2012) according to the mc3 variant calling protocol (Ellrott et al., 2018), a consensus-based gold standard approach adapted from crowdsourced benchmarking data (i.e. DREAM challenge; Ewing et al., 2015). To make somatic variants reported across tools comparable, vt (v0.5772-60f436c3; Tan et al., 2015) was used to normalize variants, decompose biallelic/block variants, sort variants and produce a final, unique list of variants for each caller. Somatic variants that were reported by more than one tool and not overlapped by any Pindel call, or that were reported by Pindel, were retained for further analysis. Germline variants were called using GATK’s HaplotypeCaller (v3.7-0-gcfedb67; McKenna et al., 2010), and we used VariantFiltration with cluster size 3 and cluster window size 15 to flag and subsequently eliminate variants with total coverage of <10.0 reads, quality by read depth of <2.0, and phred quality of <100.0. For consistency, germline variants were also processed with vt as described above. The Mbp of the genome covered was determined using bedtools genomecov (v2.26.0; Quinlan and Hall, 2010) where any base covered by a depth of at least three reads was considered covered, as this is the minimum read depth required for variant detection by SomaticSniper and VarScan 2. The coverage-adjusted mutational burden was calculated as the number of somatic mutations per Mbp of the genome covered. We employed HapCUT2 for patient-specific haplotype phasing. To do this, germline and somatic variants were combined into a single VCF using neoepiscope’s merge functionality. HapCUT2’s extractHAIRS software was run with the merged VCF and the tumor alignment file, allowing for the extraction of reads spanning indels, to produce the fragment file used with HapCUT2 to predict haplotypes. Neoepitopes were predicted for this cohort using the default settings of neoepiscope, either including germline variation and variant phasing (‘phased mode’) or using unphased somatic variants only (‘unphased mode’) in order to determine how phasing of variants might impact neoepitope predictions. The proportion of false-positive neoepitope predictions when not accounting for variant phasing was calculated as the number of neoepitopes predicted by the unphased mode but not the phased mode, divided by the total number of neoepitopes predicted by the unphased mode. The proportion of false-negative neoepitope predictions was calculated as the number of neoepitopes predicted by the phased mode but not the unphased mode, divided by the total number of neoepitopes predicted by the unphased mode.
For 90 patients (Carreno et al., 2015; Hugo et al., 2017; Snyder et al., 2014; Van Allen et al., 2015), complementary RNA sequencing (RNA-seq) data were available in addition to WES data (see Supplementary Table S1). We aligned these RNA-seq reads to the hg19 genome using STAR (v2.6.1c; Dobin et al., 2013) and searched for evidence of phasing of predicted somatic variants with germline or other somatic variants within the span of a single read pair to confirm predicted phasing by HapCUT2 and identify instances of phasing not detected by HapCUT2. If two variants were both present in at least one paired read, they were considered to be phased in RNA. If one of two variants predicted to be phased was missing from a read, this was considered evidence for lack of phasing only if (i) the variant present in the read was somatic and (ii) there were reads from that transcript that contained at least one somatic mutation, as contaminating normal tissue could explain the presence of a germline variant alone. Scripts to identify phasing of variants within RNA-seq reads are available at https://github.com/pdxgx/neoepiscope-paper under the MIT license.
2.3 Benchmarking
We compared the performance of neoepiscope with pVACseq (pvactools v1.0.7; Hundal et al., 2016) and MuPeXI (v1.2.0; Bjerregaard et al., 2017) on five melanoma patients for whom paired tumor and normal DNA-seq data were available (Bassani-Sternberg et al., 2016). Because MuPeXI was designed to work most readily with the GRCh38 genome build, we benchmarked all software using this build, which required several modifications of the approach described in Section 2.2 above. Paired tumor and normal WES reads were each aligned to the GRCh38 genome using bwa mem (v0.7.9a-r786; Li and Durbin, 2009), and GATK (v4.0.0.0; McKenna et al., 2010) was used to mark duplicates and perform base recalibration. Somatic variants were called using MuTect2 (GATK v3.5-0-g36282e4; Cibulskis et al., 2013), and germline variants were called using GATK HaplotypeCaller (v3.5-0-g36282e4), with VariantFiltration performed as described in Section 2.2. HLA types were predicted from tumor WES reads using Optitype (v1.3.1; Szolek et al., 2014). Patient-specific haplotypes were predicted using HapCUT2 (Edge et al., 2017) as described in Section 2.2, both with and without the inclusion of germline variation. For use with pVACseq and TSNAD, somatic VCFs were annotated with Variant Effect Predictor (v91.3; McLaren et al., 2016) and variants were phased using GATK’s ReadBackedPhasing according to pVACseq recommendations. Neoepitope prediction with pVACseq, MuPeXI, NeoPredPipe, TSNAD and neoepiscope was performed with the most updated versions/commits of the software available on March 4, 2019. We ran pVACseq with the phasing of somatic and germline variants, and neoepiscope in comprehensive mode. Across all tools, HLA-binding predictions were performed with netMHCpan (v4.0 for MuPeXI/neoepiscope, v2.8 for pVACSeq, which uses IEDB Tools’ netMHCpan integration; Nielsen et al., 2007) for consistency with MuPeXI’s requirements using alleles HLA-A*01:01, HLA-B*08:01 and HLA-C*07:01 (shared across four of the five patients). A uniform peptide length of nine amino acids was used for prediction in all cases, with a maximum binding score cutoff of 5×104 nM. Performance time and predicted neoepitope sequences were compared across tools (Figures 5 and 6), using four Intel Xeon E5-2697 v2 processors (when multithreading was possible), each with twelve 2.70 GHz processing cores. Scripts to reproduce the benchmarking described here are available at https://github.com/pdxgx/neoepiscope-paper under the MIT license. Predicted neoepitope sequences were compared across tools, and neoepitopes missed by any individual tool(s) were investigated to evaluate accuracy. Neoepitope sequences deriving from phased variants were considered accurate (true positives) if their phasing was confirmed by both HapCUT2 and ReadBackedPhasing; their accuracy was considered unknown wherever HapCUT2 and ReadBackedPhasing disagreed. If a neoepitope spanning the positions of two accurately phased variants was derived from only one half of the pair, that neoepitope sequence was considered to be a false positive, often accompanied by a false negative corresponding to the neoepitope sequence incorporating the pair of phased variants. The accuracy of neoepitope sequences deriving from putative RNA-editing events or mutations in unlocalized contigs was considered unknown.
2.4 Estimation of variant co-occurrence rates
To estimate patient-specific variant co-occurrence, predicted haplotypes were used to determine when somatic variants were proximal to either a germline variant or another somatic variant within the span of an MHC-II epitope (72 bp/24 aa) in the cohort described in Section 2.2. Because of variant decomposition during somatic variant calling, immediately adjacent somatic SNVs predicted to be in the same alleles were collapsed into a single multiple nucleotide variant. Nearby germline variants were counted both upstream and downstream of somatic variants, while somatic variants were only counted upstream of each other to avoid duplication. The proximity of frameshift variants and non-stop variants was considered based on strand specificity of any transcripts overlapping a somatic variant, such that only the effects of upstream frameshifts and non-stops were considered for downstream variants. Both the number of variants near a somatic variant and the minimum distance between a somatic variant and its nearby variants were recorded.
3 Results
3.1 Somatic and germline variant co-occurrence
We analyzed the intervariant distance distribution among somatic and germline variants from 375 tumor samples, employing HapCUT2 (Edge et al., 2017) for patient-specific haplotype phasing (see Materials and Methods section, Supplementary Tables S2–S4). There was an average of 4808 variants per patient (range: 178–73 351 variants). We found that variant co-occurrence increases approximately linearly with increasing nucleotide span, with an overall average of 2.55% and 0.28% of somatic variants located within a 72 bp inter-variant distance of another germline or somatic variant, respectively (Figure 3A). On a per-patient basis, as many as 4.84% and 5.71% of somatic variants co-occurred with at least one germline variant within the span of an MHC class I (≤33 bp) and class II (≤72 bp) epitope, respectively (Figure 3B). Additionally, as many 8.75% and 9.63% of somatic variants per patient co-occurred with at least one other somatic variant within the span of an MHC class I or class II epitope. For a subset of patients (n = 90), we also analyzed RNA-seq data to assess phasing across exon–exon junctions (see Materials and Methods section; Supplementary Table S5). Acknowledging that many variants may occur in genes that are not expressed in tumor samples, only 18.2% of phased somatic-germline variant pairs on average were covered by aligned RNA-seq reads and present in tumor-expressed transcripts (range: 0–53.3%). Similarly, only 42.1% of phased somatic–somatic variant pairs on average were covered by aligned RNA-seq reads and present in tumor-expressed transcripts (range: 0–100%). Among variant pairs phased across exon–exon junctions using DNA-seq data, an average of 76.1% of those covered by RNA-seq reads (228 pairs) were able to be validated by corresponding RNA-seq data, while a small fraction of those covered (2.02%) demonstrated discordant phasing. RNA-seq also identified an additional 0.17% of phased variant pairs on average across exon–exon junctions, respectively, that were not identified using DNA-seq.
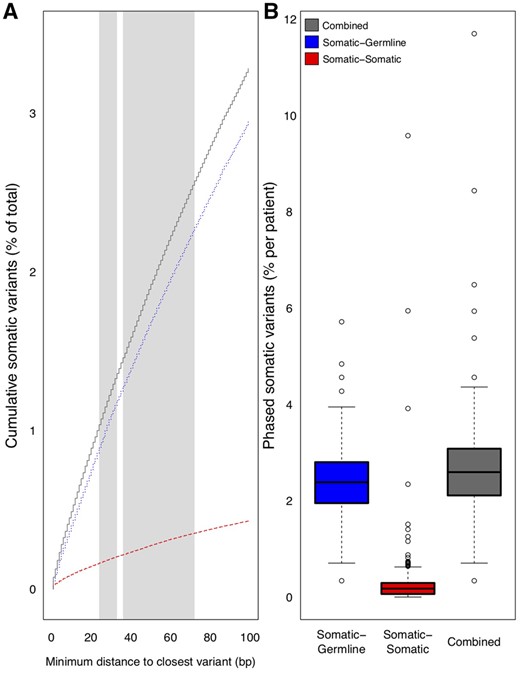
Variant co-occurrence among 285 melanoma patients, 34 NSCLC patients, and 28 colon, endometrial, and thyroid cancer patients. A) The cumulative average percentage (y-axis) of somatic variants across all tumors that co-occur with germline variants (dotted line), other somatic variants (dashed line), or either type of variant (solid line) is shown as a function of increasing nucleotide span (x-axis). Canonical MHC Class I and Class II epitope size ranges are shaded in light gray (24-33bp and 36-72bp, respectively). B) Box plots demonstrating per-patient percentage of somatic variants (y-axis) across all tumors that co-occur with germline variants (left), other somatic variants (middle), or either variant type (right)
The percentage of phased somatic variants was not significantly different between disease types (P = 0.067 by ANOVA; see Supplementary Figure S1), and did not correlate with tumor mutational burden (Pearson's product-moment correlation of 0.092, P = 0.086; see Supplementary Figure S2). Based on these data, we predicted neoepitope sequences either with or without considering variant phasing (yielding an average of 390 851 or 377 759 peptides, respectively), and found that neoepitope prediction without consideration of germline context or variant phasing produces a median of 3.82% false positive results and 4.85% false negative results (see Materials and Methods section), resulting in both an inaccurate and incomplete picture of the cancer neoepitope landscape.
3.2 Neoepitope prediction consequences of variant phasing
We identified specific examples of co-occurring variants from real patient data, as above, and show the neoepiscope-predicted consequences of both variant phasing and germline context for each of these examples in Figure 4. Co-occurrence of neighboring somatic and germline SNVs can produce neoepitopes with multiple simultaneous amino acid changes. For instance, germline (chr11:56230069 T->C) and somatic (chr11:56230065 C->T) variants in a melanoma patient [‘Pat41’ (Van Allen et al., 2015)] give rise to a collection of potential compound neoepitopes containing two adjoining amino acid changes (germline K270R and somatic M271I in ENST00000279791) (Figure 4A). Such hybrid (somatic + germline) co-occurrence within the span of an MHC Class I or Class II epitope accounts for an average of 2.55% of somatic variants overlapping transcripts (range: 0.34–5.71%).
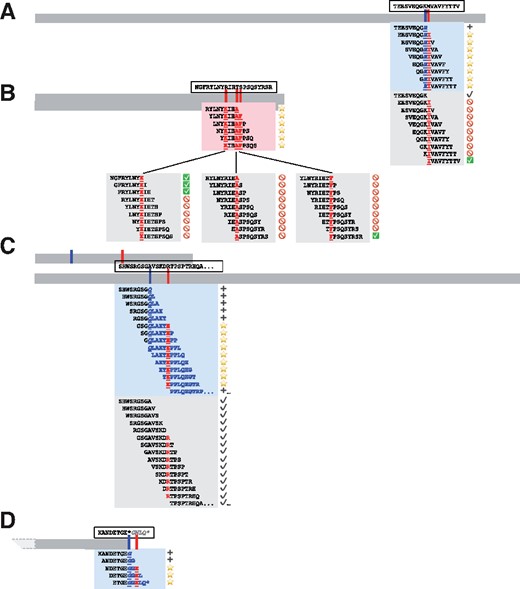
Neoepitope-level consequences of phased variants. Four coding sequences are shown to relative scale (horizontal bars), with corresponding approximate variant locations shown in tick marks for somatic and germline variants. Amino acid sequences corresponding to the reference coding sequence are shown in dashed boxes above their corresponding transcripts, with “…” indicating additional unspecified sequence, a “*” denoting a stop codon, and italicized characters indicating untranslated sequence. Highlighted boxes directly below the transcript bars contain 9mer neoepitopes and their immediate context as predicted from phased variants, with shading corresponding to somatic variants alone (part B) or including germline context (parts A, C, and D). Lower highlighted boxes contain 9mer neoepitopes and their immediate context as predicted from somatic variants in isolation, without consideration of phasing or germline context. Amino acid sequences that are directly affected by germline or somatic variants are displayed in bold underlined (somatic) or bold underlined italics (germline), while downstream variant consequences are shown without underline, and silent variant effects are neither bolded nor underlined. Corresponding symbols are shown at the right of each peptide and demonstrate the anticipated consequence of incorporating variant phasing and germline context, with stars denoting novel neoepitopes, NOT symbols denoting incorrectly predicted neoepitopes, checkboxes denoting correctly predicted neoepitopes, plus signs denoting novel proteomic context, and black check marks denoting consistent proteomic context. Co-occurring variant phenomena correspond to real patient data as noted in the text and are depicted as follows: A) germline SNV + somatic SNV, B) multiple somatic SNVs, C) germline frameshift deletion + downstream somatic SNV, and D) germline non-stop variant + downstream somatic SNV
Three neighboring somatic variants (chr21:31797833 C->T, chr21:31797825 T->C, and chr21:31797821 G->A) in another melanoma patient [‘37’ (Roh et al., 2017)] give rise, jointly, to three amino acid changes (R133K, T136A, and S137F in ENST00000390690; Figure 4B). In such cases of neighboring SNVs, the anticipated proportion of compound neoepitopes is inversely and linearly related to the distance separating neighboring variants on both sides (Figure 4B). Co-occurrence of two or more somatic variants accounts for an average of 0.28% of somatic variants overlapping transcripts (range: 0–9.63%).
It is also possible for frameshift indels to be located within upstream coding sequences near one or more phased variants. For instance, a somatic SNV (chr10:76858941 C->T) in the context of an upstream germline frameshifting deletion (chr10:76858961 CT->C) in another melanoma patient [‘Pt3’ (Hugo et al., 2017)] introduces multiple novel potential neoepitopes arising from ENST00000478873 (Figure 4C). Importantly, without germline context, this somatic SNV is predicted to be a silent mutation and would not otherwise be predicted to give rise to any neoepitopes. We analyzed the prevalence of frameshift indels neighboring downstream variants within a 94 bp window (distance corresponding to an estimated ∼95% probability of encountering a novel out-of-frame stop codon). On a per patient basis, an average of 0.36% of somatic variants overlapping transcripts were phased with an upstream frameshifting indel (range: 0–2.71%).
Finally, stop codon mutations that enable an extension of the reading frame may expose one or more downstream somatic or germline variants. For instance, a germline SNV (chr3:17131402 T->G) in another melanoma patient [‘Pt1’ (Hugo et al., 2017)] eliminates a stop codon in ENST00000418129, with an anticipated extension of translation of an additional 5 amino acids prior to encountering the next stop codon. However, the co-occurrence of a downstream somatic SNV (chr3:17131406 G->A) alters the anticipated peptide composition (Figure 4D). We analyzed the frequency of such non-stop variants co-occurring with one or more downstream variants within a 94 bp window (distance corresponding to an estimated ∼95% probability of encountering a novel out-of-frame stop codon). On a per patient basis, an average of 0.003% of somatic variants overlapping transcripts were phased with a non-stop variant (range: 0–0.11%).
3.3 Software performance and benchmarking
We attempted to compare the performance of neoepiscope with all other publicly available neoepitope prediction tools but were unable to recapitulate several of these pipelines despite the availability of the code. For example, the local version of CloudNeo had several Dockerfile and code-level errors that prevented us from running the installed tool. This raises a significant reproducibility and generalizability issue among many existing software tools. However, we were able to successfully benchmark four popular neoepitope prediction tools (pVACseq, MuPeXI, NeoPredPipe and TSNAD) along with neoepiscope.
Run as a full pipeline, neoepiscope outperformed pVACseq by an average of 44.7 min per sample and outperformed MuPeXI by 3.4 min per sample, even when including germline variant calling (see Figure 5; Supplementary Table S6). NeoPredPipe and TSNAD outperformed neoepiscope by an average of 79.5, and 74.9 min per sample, respectively. These differences were predominantly accounted for by the average 76.7 min per sample runtime of germline variant calling that is required prior to running neoepiscope, as well as the additional 1.5 min per sample runtime of HapCUT2 when phasing somatic and germline variants. Excluding any preprocessing steps, neoepiscope outperformed pVACseq, MuPeXI and TSNAD by 38.8, 4.86 and 3.22 min, respectively, and was outperformed by NeoPredPipe by 1.37 min.
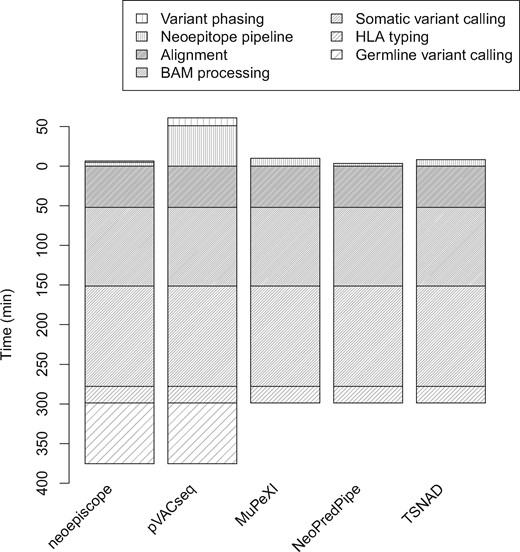
Performance benchmarking of neoepiscope, pVACseq, MuPeXI, NeoPredPipe and TSNAD. The run time (y-axis) of neoepiscope including germline variation was compared with those of pVACseq, MuPeXI, NeoPredPipe and TSNAD at a pipeline level. Time zero indicates the beginning of the neoepitope calling process from somatic (and germline, if relevant) variants, such that steps in the pipeline prior to neoepitope prediction (e.g. sequence read alignment, somatic variant calling, etc.) are displayed below time zero, and steps during or required for neoepitope calling are displayed above time zero. Only neoepiscope and pVACseq pipelines required variant phasing. Variant Effect Predictor (VEP) runtimes were included in the pVACseq and TSNAD neoepitope prediction steps (VEP is a prerequisite for pVACseq and TSNAD)
We also compared the neoepitope sequences predicted by each tool (see Supplementary Table S7). Across all tools, 35 858 peptide sequences were predicted. Although the majority of neoepitopes predicted across all tools (55.0% or 19 727 peptides) is consistent across all prediction tools, there are significant nuances and differences in predictions between tools (Figure 6). Of concern, we find that 22.9% of total neoepitopes (8225 peptides) are missed by TSNAD due to incomplete transcript/variant annotation, while 0.6% (207 peptides) are erroneously reported due to incorrect variant interpretation. NeoPredPipe faces similar issues, with 4.9% of total neoepitopes (1751 peptides) missed due to incomplete annotation, and an additional 14.8% of neoepitopes (5319 peptides) resulting from incorrect variant interpretation. MuPeXI also reports 0.3% of neoepitopes (90 peptides) due to incorrect variant interpretation. Among neoepitope prediction tools, only neoepiscope, MuPeXI and NeoPredPipe are able to process peptide sequences resulting from a disrupted stop codon. For instance, pVACseq, TSNAD and NeoPredPipe are unable to identify the 0.08% of total neoepitopes (29 peptides) resulting from frameshift variants that extend the open reading frame past a stop codon. Similarly, pVACseq, TSNAD and MuPeXI are unable to identify the 0.01% of neoepitopes (five peptides) resulting from stop codon point mutations.
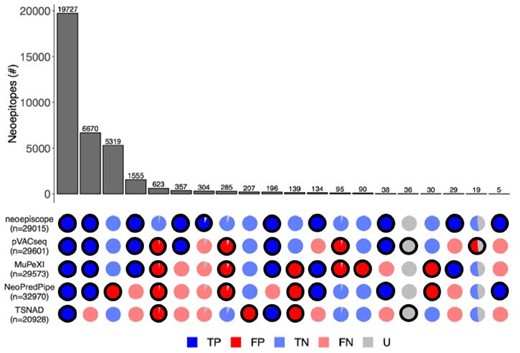
Detailed comparison of the complete set of neoepitope predictions from neoepiscope, MuPeXI, pVACseq, NeoPredPipe and TSNAD. Patterns of agreement or disagreement among groups of neoepitopes predicted by different combinations of tools across five melanoma patients are shown along each column (e.g. the first column corresponds to neoepitopes predicted by all tools). Each row indicates the neoepitope predictions associated with the indicated tool, with the total number of neoepitopes predicted by each tool shown as n. The number of neoepitopes in each column (bar in upper pane) corresponds to the size of the subset predicted by the indicated combination of tools (outlined circles in the bottom pane). The veracity of predictions corresponding to each group of neoepitopes are shown as pie charts, with colors corresponding to true positive (‘TP’, dark blue), false positive (‘FP’, dark red), true negative (‘TN’, light blue), false negative (‘FN’, light red) and uncertain (‘U’, gray) predictions. Uncertain predictions are considered a result of one or more factors including origin from unassembled contig regions, the presence of RNA edits or inconsistencies in variant phasing predictions between HapCUT2 and GATK’s ReadBackedPhasing
Overall, 1% of predicted neoepitopes (357 peptides) result from phased variants and are predicted by both neoepiscope and pVACseq but not other tools. A similarly sized set (0.8% or 304 peptides) of neoepitopes resulting from phased variants is identified by neoepiscope alone (Supplementary Table S8). Of these, 24 peptides (7.9%) appear to be due to inconsistencies in variant phasing between HapCUT2 and GATK ReadBackedPhasing; these neoepitopes are flagged as uncertain. However, the other 92.1% of these neoepitopes (280 peptides) are phased consistently by both HapCUT2 and GATK ReadBackedPhasing but are incorrectly missing from pVACseq output. Conversely, 3.3% of all neoepitopes (1172 peptides) are incorrectly reported (false positives), as they are predicted to be absent when taking variant phasing into account. Of these, 3.4% (40 peptides) appear to be due to inconsistencies in variant phasing between HapCUT2 and GATK ReadBackedPhasing and are flagged as uncertain. However, the remaining 96.6% (1132 peptides) are consistently phased by both HapCUT2 and GATK ReadBackedPhasing but erroneously reported by pVACseq and all other tools with the exception of neoepiscope. Taken together, a total of ∼6% of all neoepitopes (∼2151 peptides) are incorrectly or incompletely predicted without appropriately accounting for variant phasing.
There are several other anticipated discrepancies among neoepitope prediction tools. For instance, there were nine neoepitopes (0.03%) derived from a mutation to a transcript from an unlocalized contig (chr1_KI270711v1_random) reported only by pVACseq and 36 (0.1%) associated with presumed RNA-editing events (on transcripts ENST00000361453 and ENST00000362079) which were reported only by pVACseq and TSNAD due to discrepancies in genomic annotation and variant effect prediction, respectively. However, in the absence of RNA-seq data, there is no way to reasonably differentiate performance for these sites. For the purposes of this analysis, we considered neoepitopes arising from nonsense-mediated decay, polymorphic pseudogene sequences, IG V and TR V transcripts to be bonafide sources of antigenic peptides (Apcher et al., 2011); however neoepiscope can be parameterized to include or exclude any of these sequences as desired for a more nuanced interpretation of variant effects on putative neoepitopes.
4 Discussion
We report here a novel, performant and flexible pipeline for neoepitope prediction from DNA-seq data (neoepiscope), which we demonstrate improves upon existing pipelines in several ways. In particular, we demonstrate the importance of germline context and variant phasing for accurate and comprehensive neoepitope identification. Not only do co-occurring somatic variants represent a source of previously unappreciated neoepitopes, but also these compound epitopes may harbor more immunogenic potential compared with peptides from isolated somatic variants (due to multiple amino acid changes within the same epitope; Pradeu and Carosella, 2006; Yadav et al., 2014). Furthermore, we find that neoepiscope improves both sensitivity and specificity compared with existing software [which incorrectly or incompletely predicts ∼5% of neoepitopes (see Materials and Methods section)]. Indeed, this study is the first to critically evaluate and benchmark multiple neoepitope prediction tools, and raises the specter of broad reproducibility, accuracy and usability issues within the field. Finally, we note that the neoepiscope framework is sufficiently flexible to accommodate numerous variant types, nonsense-mediated decay products and epitope prediction across different genomes.
There are several limitations to neoepiscope at present. For instance, the software does not currently leverage RNA-seq data for gene expression or transcript-level variant phasing (Castel et al., 2016). Although the importance of gene dosing for neoepitope presentation remains unclear at this time, the incorporation of transcript-level variant quantification could potentially improve the accuracy of neoepitope prediction. In addition, neoepiscope does not currently predict neoepitopes that may arise from alternative splicing events, RNA-editing, structural variation or gene fusions which are common across multiple cancer types (Kahles et al., 2018; Smart et al., 2018; Xu et al., 2018; Zhang et al., 2017). Because neoepiscope implicitly assumes a diploid genome when using HapCUT2 for variant phasing, it may have decreased predictive accuracy in the context of significant somatic copy number alterations, though we do not observe this to be the case in the datasets presented herein (Zack et al., 2013). In addition, the software does not account for the potentiation of the nonsense-mediated decay pathway by frameshifting indels, leading to uncertain implications for downstream neoepitope presentation in such cases. Lastly, as there remains significant uncertainty in predicting antigenicity in vivo, neoepiscope does not attempt to prioritize or rank putative neoepitopes in any way, relying instead on the end-user to interpret results according to their own preferred schema.
In the future, we will address these limitations by incorporating downstream predictive methodologies which could aid in identifying the most immunogenic candidates [e.g. for personalized vaccine development (Ott et al., 2017; Sahin et al., 2017)] and incorporating predictive features from RNA-seq as above to expand the repertoire of predicted neoepitopes. Given the proliferation of predictive tools, and their notable caveats and limitations as described in this study, we believe the field must advocate for the highest of standards in software reproducibility and transparency. Finally, we assert that future analyses should account for both germline context and variant phasing, and are happy to provide neoepiscope as a resource for the community, accordingly.
Acknowledgments
We would like to thank Mihir Paralkar and Mayur Paralkar for their support at neoepiscope’s inception. We would also like to thank Dr. Rachel Karchin for her group’s support with MHCnuggets integration. We finally thank Julianne David for her critical review of the manuscript.
Funding
This work was supported by the Sunlin & Priscilla Chou Foundation to R.F.T.
Disclaimer: The contents do not represent the views of the U.S. Department of Veterans Affairs or the US government.
Conflict of Interest: None declared.
References
1000 Genomes Project Consortium;
cancerit. (n.d.) Cancerit/dockstore-Cgpmap. GitHub. https://github.com/cancerit/dockstore-cgpmap (12 September 2018, date last accessed).
gt. (n.d.) gt1/biobambam2. GitHub. https://github.com/gt1/biobambam2 (12 September 2018, date last accessed).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}