





Abstract
Protein structure modeling can be improved by the use of distance constraints between amino acid residues, provided such data reflects—at least partially—the native tertiary structure of the target system. In fact, only a small subset of the native contact map is necessary to successfully drive the model conformational search, so one important goal is to obtain the set of constraints with the highest true-positive rate, lowest redundancy and greatest amount of information. In this work, we introduce a constraint evaluation and selection method based on the point-biserial correlation coefficient, which utilizes structural information from an ensemble of models to indirectly measure the power of each constraint in biasing the conformational search toward consensus structures.
Residue contact maps obtained by direct coupling analysis are systematically improved by means of discriminant analysis, reaching in some cases accuracies often seen only in modern deep-learning-based approaches. When combined with an iterative modeling workflow, the proposed constraint classification optimizes the selection of the constraint set and maximizes the probability of obtaining successful models. The use of discriminant analysis for the valorization of the information of constraint datasets is a general concept with possible applications to other constraint types and modeling problems.
MSA for the targets in this work is available on https://github.com/m3g/2021_Bottino_Biserial. Modeling data supporting the findings of this study was generated at the Center for Computing in Engineering and Sciences, and is available from the corresponding author LM on request.
Supplementary data are available at Bioinformatics online.
1 Introduction
Dealing with noisy and incomplete datasets is a central problem for the field of biomolecular modeling, especially in the case of protein structure determination. The most successful strategies developed over the last decades in this field share a common paradigm of utilizing target-specific constraints to customize energy functions and restrain the conformational search, improving modeling output results (Abriata et al., 2019; Kinch et al., 2016; Schaarschmidt et al., 2018; Taylor et al., 2014). It is reasonable to say, therefore, that the extent of those strategies’ success depends not only on obtaining or predicting good constraints, but also from sorting or selecting the best constraints from every knowledge source, the accuracy and availability of which can be heterogeneous and hard to estimate.
State-of-art protocols for constraint map estimation rely heavily on data obtained from amino acid coevolution analysis. This approach has evolved greatly over the last decades from a simple computation of correlated substitutions (Göbel et al., 1994) on multiple sequence alignments to a myriad of different methods (de Juan et al., 2013) accompanied by a history of successful employment evidence (Huang et al., 2016; Ovchinnikov et al., 2015, 2017).
Among those evolutionary methods, the most successful ones to date rely on applying Machine Learning techniques (Kandathil et al., 2019b). Some of them predict continuous pairwise distance distributions, to which is attributed part of the leap of progress in template-free modeling witnessed on recent CASP competitions (Kryshtafovych et al., 2019; Service and Service, 2020). More recently, the prediction of continuous values for other structural features like backbone torsion angles or inter-residue orientations has also been explored by these cutting edge methods (Billings et al., 2019; Senior et al., 2020; Wang et al., 2017; Yang et al., 2020).
Many of the popular protocols rely on the prediction of binary contact maps (Adhikari, 2020; Adhikari et al., 2018; Dos Santos et al., 2019, 2018; Hopf et al., 2019; Jones et al., 2012; Kaján et al., 2014; Kandathil et al., 2019a; Marks et al., 2011; Morcos et al., 2011; Ovchinnikov et al., 2014; Seemayer et al., 2014), from which bounded restraining potentials are derived. Predicted maps sometimes lack completeness and may contain a significative number of incorrect contacts, leading to inaccurate sampling of the conformational space, as illustrated in CASP13 contact predictions, where roughly 1/3 of groups achieved over 0.5 precision, with a maximum around 0.7 (Shrestha et al., 2019). Since it is widely recognized that only a small portion of the true contact map of a protein is necessary for a successful modeling (Kim et al., 2014; Mandalaparthy et al., 2018; Skolnick et al., 1997), protein structural modeling from this kind of data can be thought as the problem of adequately selecting the best constraint subset from the estimated map.
Combinatorial variable or feature selection techniques are widely spread amid fields employing multivariate data analysis, but this kind of approach is prohibitive for biomolecular modeling due to the computational cost of evaluating multiple subsets. Hence, a guided selection criterion, in the shape of a feature score tailored to utilize information pertinent to the modeling output itself, might be the solution to this limitation.
Constraint selection strategies specific to protein modeling are not entirely new, but have historically pertained to the field of NMR-assisted modeling, particularly for the purpose of dealing with ambiguous constraints during the conformational search (Brünger et al., 1998; Nilges, 1995; Rieping et al., 2005, 2007). Here, we develop a flexible approach to constraint selection, agnostic to the type and origin of the constraints, and which can easily be coupled with different modeling workflows.
We propose a constraint scoring strategy for binary contacts based on the ability of each constraint to discriminate model quality as assessed by consensus modeling scores or other quality measures. Instead of a simple variance measure, however, the proposed score—point-biserial correlation coefficient—incorporates a structure as reference, causing the selected constraint subset to indirectly bias the sampled conformational space toward the vicinity of such reference.
In summary, we adopt an iterative modeling strategy, based on the steered stepwise valorization of a preliminary constraint set. After each iteration of model generation, a reference structure is selected by a quality assessment method and, for each possible constraint, we compute its ability to differentiate between models similar or dissimilar to such reference. The constraints positively correlated with model quality are selected for a subsequent round of structure modeling. Given the reference model has adequate fold accuracy, this strategy leads to higher constraint true-positive rates and better model ensembles. Even though this work is focused on the rescoring of Direct Coupling Analysis constraints through an incremental fragment-based modeling strategy, the principle of structural discrimination is rather universal and may find application in other biomolecular modeling protocols.
2 Approach
Strategies to predict amino acid residue contacts of proteins commonly provide a list of predicted contacts and some score reflecting the accuracy of the estimate. These scores are often employed to rank the constraints to be employed on modeling. This decision criterion is not optimal for protein structure modeling: The set of contacts which provide the best modeling is not necessarily that which is predicted with greater accuracy, even assuming that the constraints are correct. In particular, amino acids that are close in the primary sequence will very likely be close in the tertiary structure and display large prediction scores. Second, short primary range are under-performant (in a modeling sense) when compared to medium and long range contacts (Mandalaparthy et al., 2018), especially because they only encode local structural information (Censoni and Martínez, 2018).
To circumvent this issue, many contact estimation methods will employ some kind of cutoff for the separation between residues in the primary sequence, as an attempt to filter out trivial constraints a priori. Although this is generally a good enough solution, we understand that this arbitrary triviality should be re-evaluated after every model generation step in order to respect the particularities of each modeling strategy and incorporate structural features derived from the very models generated. This conceptual approach is valid not only for contacts within residues close in the primary sequence, but also in general: Given a particular protein and modeling strategy, some contacts might be useful constraints for conformational search, and others might not. Incorrect contact predictions of course must be avoided. Furthermore, some correct constraints might be detrimental for conformational search by frustrating the energy surface too early given the search protocol of choice. The optimal constraint set for energy minimization and conformational search is, thus, specific for each structure and modeling protocol.
In particular, the structurally trivial—to be avoided—constraints are those that often combine the following characteristics: they are obeyed with high frequency even on unconstrained modeling results, show redundant or sometimes even repeated information and have a high degree of structural localization, being often observed near the protein terminals or over the same sections of continuous secondary structure.
From definition, it is understandable that a large portion of the trivial constraints are of short primary range, but that is not always the case: it is possible that a medium or long-range constraint arises naturally during modeling (for example, on fragment-based approaches); in contrast, some short-range constraints could happen over loops and be important for defining local shapes on hard targets when there was low availability of evolutionary information. In light of such observations, we defend that a candidate constraint scoring measure must be able to naturally (without supervision or arbitrary cutoffs) filter out trivial constraints from the recovered set.
3 Materials and methods
3.1 Target list and coevolution analysis
A target is a protein to be modeled, given its primary sequence. In this work, we selected nine different proteins from both α and α-β fold classes, with sequence lengths (L) between 71 and 167, averaging 130 amino acids. For each protein, the first step is the identification of a family and generation of a multiple sequence alignment containing the target sequence. MSAs for the whole protein family of those targets were obtained from the respective Pfam entries (El-Gebali et al., 2019), identified by the InterProScan (Jones et al., 2014) search engine. Clustal Omega (Sievers et al., 2011; Sievers and Higgins, 2018) was utilized to align the target sequence to the family profile. Information regarding target length, MSA size and diversity is portrayed on Table 1.
Target information for the constraint selection experiments
| Target | Sequence | CATH | MSA | MSA | Pfam | Reference |
|---|---|---|---|---|---|---|
| PDBID_CHAIN | Length (L) | fold class | Size (N) | Diversity (Neff)a | Family ID | |
| 1C52_A | 131 | α | 27780 | 9.19 | PF00034 | Than et al. (1997) |
| 1C75_A | 71 | α | 26546 | 7.43 | PF13442 | Benini et al. (2000) |
| 1D06_A | 130 | α-β | 40030 | 8.38 | PF00989 | Miyatake et al. (2000) |
| 1E6K_A | 130 | α-β | 78398 | 5.37 | PF00072 | Solà et al. (2000) |
| 1KAO_A | 167 | α-β | 62267 | 5.29 | PF00071 | Cherfils (1997) |
| 1RQM_A | 105 | α-β | 55298 | 6.97 | PF00085 | Leone et al. (2004) |
| 4LE1_A | 139 | α-β | 78398 | 7.18 | PF00072 | Trajtenberg et al. (2014) |
| 5CXO_B | 134 | α-β | 21409 | 9.48 | PF12680 | Luhavaya et al. (2015) |
| 5P21_A | 166 | α-β | 62267 | 5.44 | PF00071 | Pai et al. (1990) |
| Target | Sequence | CATH | MSA | MSA | Pfam | Reference |
|---|---|---|---|---|---|---|
| PDBID_CHAIN | Length (L) | fold class | Size (N) | Diversity (Neff)a | Family ID | |
| 1C52_A | 131 | α | 27780 | 9.19 | PF00034 | Than et al. (1997) |
| 1C75_A | 71 | α | 26546 | 7.43 | PF13442 | Benini et al. (2000) |
| 1D06_A | 130 | α-β | 40030 | 8.38 | PF00989 | Miyatake et al. (2000) |
| 1E6K_A | 130 | α-β | 78398 | 5.37 | PF00072 | Solà et al. (2000) |
| 1KAO_A | 167 | α-β | 62267 | 5.29 | PF00071 | Cherfils (1997) |
| 1RQM_A | 105 | α-β | 55298 | 6.97 | PF00085 | Leone et al. (2004) |
| 4LE1_A | 139 | α-β | 78398 | 7.18 | PF00072 | Trajtenberg et al. (2014) |
| 5CXO_B | 134 | α-β | 21409 | 9.48 | PF12680 | Luhavaya et al. (2015) |
| 5P21_A | 166 | α-β | 62267 | 5.44 | PF00071 | Pai et al. (1990) |
Neff was measured by the exponential of entropy, averaged over all columns of the L-length MSA (Peng and Xu, 2010).
Target information for the constraint selection experiments
| Target | Sequence | CATH | MSA | MSA | Pfam | Reference |
|---|---|---|---|---|---|---|
| PDBID_CHAIN | Length (L) | fold class | Size (N) | Diversity (Neff)a | Family ID | |
| 1C52_A | 131 | α | 27780 | 9.19 | PF00034 | Than et al. (1997) |
| 1C75_A | 71 | α | 26546 | 7.43 | PF13442 | Benini et al. (2000) |
| 1D06_A | 130 | α-β | 40030 | 8.38 | PF00989 | Miyatake et al. (2000) |
| 1E6K_A | 130 | α-β | 78398 | 5.37 | PF00072 | Solà et al. (2000) |
| 1KAO_A | 167 | α-β | 62267 | 5.29 | PF00071 | Cherfils (1997) |
| 1RQM_A | 105 | α-β | 55298 | 6.97 | PF00085 | Leone et al. (2004) |
| 4LE1_A | 139 | α-β | 78398 | 7.18 | PF00072 | Trajtenberg et al. (2014) |
| 5CXO_B | 134 | α-β | 21409 | 9.48 | PF12680 | Luhavaya et al. (2015) |
| 5P21_A | 166 | α-β | 62267 | 5.44 | PF00071 | Pai et al. (1990) |
| Target | Sequence | CATH | MSA | MSA | Pfam | Reference |
|---|---|---|---|---|---|---|
| PDBID_CHAIN | Length (L) | fold class | Size (N) | Diversity (Neff)a | Family ID | |
| 1C52_A | 131 | α | 27780 | 9.19 | PF00034 | Than et al. (1997) |
| 1C75_A | 71 | α | 26546 | 7.43 | PF13442 | Benini et al. (2000) |
| 1D06_A | 130 | α-β | 40030 | 8.38 | PF00989 | Miyatake et al. (2000) |
| 1E6K_A | 130 | α-β | 78398 | 5.37 | PF00072 | Solà et al. (2000) |
| 1KAO_A | 167 | α-β | 62267 | 5.29 | PF00071 | Cherfils (1997) |
| 1RQM_A | 105 | α-β | 55298 | 6.97 | PF00085 | Leone et al. (2004) |
| 4LE1_A | 139 | α-β | 78398 | 7.18 | PF00072 | Trajtenberg et al. (2014) |
| 5CXO_B | 134 | α-β | 21409 | 9.48 | PF12680 | Luhavaya et al. (2015) |
| 5P21_A | 166 | α-β | 62267 | 5.44 | PF00071 | Pai et al. (1990) |
Neff was measured by the exponential of entropy, averaged over all columns of the L-length MSA (Peng and Xu, 2010).
Direct-coupling analysis (Morcos et al., 2011) was performed on the resulting MSA utilizing the GaussDCA (Baldassi et al., 2014) protocol, which provided Direct Information (DI) values for each pair of amino acids on the target primary structure. The residue pairs were sorted through descending values of DI, generating a list from which the top L (L being the primary sequence length) pairs are selected as the preliminary estimated contacts and encoded as constraints for modeling.
We acknowledge that the modeling result will depend on the target size, Neff, constraint estimation protocol and modeling strategy. Upon selecting our targets, we intentionally approached smaller sizes of domain length, while also avoiding the harder-to-model beta folding class. This kept us away from the size limitations of fragment-based ab initio folding. In our targets, DCA constraints were not good enough to definitely guide modeling toward fold-quality models without a strongly positively biased fragment library (more comments on Supplementary Section S1). This provided room for improvement, allowing our selection strategy to increase the value of noisy and insufficient constraint sets. Nevertheless, the principle of using discrimination analysis to enrich the constraint set is universal, as exemplified by an additional target modeled with a deep-learning-estimated contact set, available in Supplementary Section S2.
3.2 Contact definition and constraining potential
Constraints were incorporated with bounded flat-bottom potentials between Cβ atoms of both residues (except for glycines, where the Cα atom was utilized). Along the flat portion, this potential assumes the value of zero, growing quadratically with the amount of violation when the boundaries are trespassed. In agreement with contemporary contact definitions (Shrestha et al., 2019), the upper boundary of the flat portion or maximum separation distance between atoms in contact was 8.0 Å. A minimum separation distance (lower boundary) of 3.5 Å was also added in order to reinforce the prevention of steric clash between the centroid atoms in the coarse-grained ab initio phase of modeling.
3.3 Modeling experiments
For each target, we performed six types of modeling experiments. The first of them, which we called DCA_INIT (1), is a preliminary modeling round that employs the top L contacts derived from the DCA-based coevolution analysis. This round is important because it provides not only a baseline of modeling performance for our selection protocol, but also an initial pool of structures to be analyzed and over which the structural discrimination selection protocol can act. To maintain consistency, in every modeling round for every target, the number of models generated is equals to 10*L. All our modeling efforts were performed with the Rosetta abinitiorelax framework (Raman et al., 2009) with a homologous-free fragment library obtained via the Robetta server (Kim et al., 2004). Configuration files, when pertinent, are included on an open repository, along with a reproducible example of our analysis (see end of Section 3.3).
From this first modeling round, we blindly elect a representative model via a simple consensus score inspired based on the Davis‐QAconsensus (Kryshtafovych et al., 2014), and perform two other experiments: the first one, employing constraint selection through the point-biserial correlation coefficient through three incremental rounds, named BIS_CONS (2) and a separate modeling round, based on naive random resampling of contacts on this very consensus model, named L_CONS (3). On the BIS_CONS experiment, all residue pairs are rescored through the value of rpb and contacts are ranked again, generating a new list with the top L contacts for input on the next rounds. In the L_CONS experiment, each model is generated from a random set of L contacts consistent with the consensus reference elected after the DCA_INIT round.
Another branch of experiments also derives from the DCA_INIT starting point, in an effort to decouple the reference model selection strategy from the constraint selection strategy. For this branch, the reference model is selected not by a consensus strategy, but by an idealized selector which always picks the model with highest TM-score (Zhang and Skolnick, 2004) against the native structure, which can be called the ‘best model’. Again, in this branch, two experiments were performed: one with constraint selection using the value of rpb, named BIS_BEST (4); another with naive resampling of the contacts found on the best model, named L_BEST (5). Finally, an upper-boundary control experiment was also performed, with random sampling of native contacts, where every constraint is a true-positive on the crystallographic structure. This final experiment was named L_CRYS (6).
At the end of each round, the models are added to the existing model pool, incrementing its size and contributing new information. This analysis-modeling-incrementing block is then iterated according to experimental design. Inside each branch of experiments and after each modeling round, the whole model pool undergoes all-on-all structural alignment with the LovoAlign software (Martínez et al., 2007) to update the elected representative. For the targets in this work, we performed three rounds of this incremental block on the BIS experiments. Figure 1 shows a visual summary of the methodology.
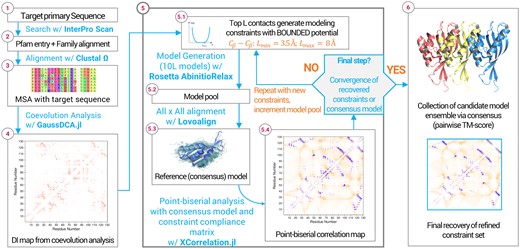
Visual summary of the methodology. The central rectangle highlights the incremental modeling rounds with point-biserial coefficient acting as the main tool for constraint selection, rescoring all possible constraints through their ability to discriminate toward the reference consensus model
To perform the constraint selection on the BIS rounds, we build a binary contact constraint-compliance matrix for every model, and the point-biserial correlation of each constraint is evaluated using TM-score as the continuous classification variable of the similarity of each model to the consensus reference. Contacts are sorted through this score, generating a new constraint set of L contacts which serve as input to a new modeling round. A minimal reproducible example for one of the targets with detailed instructions, codes and configuration files, as well as the selection scripts and production parameters for the abinitio protocol is available at https://github.com/m3g/2021_Bottino_Biserial.
3.4 Point-biserial correlation coefficient as constraint selection criterion
The point-biserial correlation coefficient (rpb) (Pearson, 1900) is a correlation coefficient with properties which adapt to the problem of constraint selection. It is sometimes regarded as the dichotomic case of the traditional correlation coefficient derived by the same author and provides a measure of discrimination between a binary variable—the observance or violation of a given constraint throughout protein models—and a continuous variable, in our case the estimated structural quality of models.
For a long time, this coefficient has been employed on large-scale educational assessment, in order to estimate correlations between the binary answer to a question from a test and the student’s score or ability: questions with higher point-biserial coefficients are the most discriminatory between students of high and low ability. It is not only a tool of test item validation, but also exam refinement, since it is possible to improve the performance of an exam by excluding test items with low or negative values of rpb (LeBlanc and Cox, 2017). This selection strategy is traditionally established as allowing for better predictive performance than simply selecting the hardest items—those consistent with the most performant students.
Our idea is to adapt this concept to constraint-assisted biomolecular modeling, in such a way that the aim is not to find out constraints consistent with the best models; actually, we want to find out which constraints are most discriminating between the worst and best models, given a consensual reference, employing rpb as a measure for such discriminating power.
Point-biserial correlation has a number of properties of interest to our approach. Being a correlation coefficient, its value will always belong to the closed interval , providing not only a convenient natural sign change at , but also an interesting value range for input at statistical learning models, which often require some sort of scaling or normalization. Another interesting feature is that, thanks to the normalization factor , constraints with high frequency ( small)—such as most structurally trivial constraints—experience a heavy penalization on the absolute value of their point-biserial score.
It is important to mention that the same principle of finding zero-order correlations between individual constraints and model qualities could also be applied in workflows that use predicted continuous distances instead of binary contacts. The type of correlation itself would have to be changed, since point-biserial demands a dichotomous variable for the constraint compliance axis, but other types of correlation could be coupled with adapted non-discrete metrics of constraint violation, following a similar principle.
4 Results and discussion
Individualized results for all six experiments and all nine targets can be collectively discussed in light of a representative example. For this discussion, we elected PDB_1D06_A as such an example, portraying the relevant data in Figure 2. The same data regarding all other targets can be found in Supplementary Figures SF1.1 through SF1.8, and additional discussions are available in supporting text ST1.
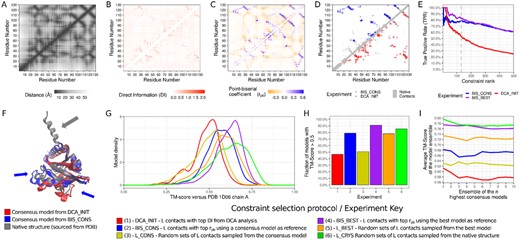
Modeling results for representative target PDB_1D06_A. (A) Native distance map for target crystallographic structure. (B) Estimated DI map from DCA analysis on Target family MSA. (C) Rescored point-biserial correlation map from BIS_CONS round. (D) Selected constraints for DCA_INIT and BIS_CONS modeling experiments. (E) Cumulative True Positive Rates for sorted constraints in the DCA_INIT, BIS_CONS and BIS_BEST experiments. (F) Structural alignment of the target native structure with the consensus models elected after DCA_INIT and BIS_CONS rounds. Blue arrows indicate regions where topology improved after constraint selection; Gray arrows indicate regions wrongly sampled by reasons discussed in Section 4.3. (G) Distribution of model qualities measured through TM-score against crystallographic structure for each modeling experiment. (H) Proportion of models with correct topology (TM-score of alignment with native structure > 0.5) for each experiment. (I) Average TM-score of the ensemble with n models recovered after the last round of each modeling experiment
4.1 Structural information improves sparsity and structural motifs of contact maps
Figure 2A represents the native distance map of the target crystallographic structure, in which darker areas represent closer residues. Inspection of the preliminary estimated DCA maps in Figure 2B shows that some features of the native distance map are captured in the coevolution analysis, but evolutionary contacts are generally sparse (with highlights to some undersampled regions between residues 70 and 110) and noisy, with little definition of characteristic secondary-structure motifs near the diagonal of the DI (Direct Information) map. When inspecting the BIS map (Fig. 2C), however, we note a visual improvement of definition on the structural features as a result of the incorporation of structural information, specifically some well-defined beta strands and the rise of important constraints in undersampled regions of the DI map. This result was consistently observed for all targets evaluated in this work.
4.2 Point-biserial correlation gives high scores to true-positive constraints of larger primary separation
The distribution of selected constraints on the residue pair map (Fig. 2D) shows that the BIS contacts are better distributed through the different regions of the native map. BIS contacts appear to explore better medium and long-range true-positive regions, while also being more cohesive at that matter. Part of this claim is confirmed by Figure 2E, which shows the accumulated true-positive rate along the ranked variable, for the DCA set and two BIS sets from each modeling branch (BIS_CONS and BIS_BEST). Except for some fluctuations on the very beginning of the charts, those proportions of true positives show that the BIS sets are not only superior on the L-length subset, but in almost every case the TPR for L/10, L/5 and L/2 is also greater. Additionally, the average separation of the residue pairs in the set was increased from 29 to 43 residues. We conclude that the true-positive rate and average range of the BIS sets consistently surpass that of the DCA sets, as should be expected for any constraint selection method.
4.3 Model topologies are improved after point-biserial selection
In Figure 2F, we have the opportunity to look at the 3D alignment of blindly elected models on the DCA_INIT and BIS_CONS experiment, against the crystallographic structure. In almost every target, it is possible to visually identify one or more regions where the BIS-generated model shows an important improvement on its topology, compared to the preliminary counterpart. Specifically on target PDB_1D06_A, we can identify two major regions where the correct orientation and secondary structure of the backbone were reached after the constraint selection. It is also notable that, in some targets including PDB_1D06_A, there were portions of the native structure that were incorrectly sampled in both experiments (before and after constraint selection). Closer inspection allows us to conclude that in most cases this setback was a consequence of mistakes in the secondary structure or—in the case of PDB-1D06 - the privileging of smaller gyration radius by the force-field.
Looking at quality distributions for each experiment (Fig. 2G), it can be seen that the BIS experiments tend to form taller and narrower peaks than their naive counterparts. For some targets, the peak of the BIS_CONS and BIS_BEST curves matched secondary peaks or shoulders on the curves of the DCA_INIT curve, giving the idea that the constraint selection contributed as a biasing force toward privileging quality ranges that were already sampled in previous stages.
Figure 2H portrays the fraction of generated models in the final round of each experiment which attained a TM-score larger than 0.5 relative to the crystallographic structure, which is considered to be a common standard for matching topology between protein structures (Xu and Zhang, 2010). This general pattern where the greater proportions of correct models are achieved by the BIS_CONS and BIS_BEST sets emerged not only on PDB_1D06_A, but also in most of the other targets (as shown in Supplementary Figs SF1.1–SF1.8). The fact that these fractions for the BIS sets were not only larger than their combinatorial counterparts, but also larger than the control L_CRYS experiment, gave rise to interesting conclusions discussed further in the results summary (Section 4.5).
An ensemble of 10 representative models, symbolizing the result of the modeling effort for each target, was extracted from each experiment, employing again a simple consensus strategy. The average quality of the representative ensemble (TM-score against the native structure) as a function of its size is depicted on Figure 2I. The clear pattern that emerges is consistent with our hypothesis: in the lowest end, we can find the quality for the preliminary DCA_INIT set. It is followed by the qualities in the first branch of experiments (L_CONS and BIS_CONS), where a consensus reference was chosen, where the employment of point-biserial correlations yielded better results than a naive use of the reference model. Next, we can find those pertaining to the second branch of experiments (L_BEST and BIS_BEST), where the reference model was the best one available. Again, the result for the employment of point-biserial coefficients through the best model is better than the naive use of the best model. Finally, the highest curve is occupied by the theoretical LCRYS control experiment. These observations show that the BIS sets result not only in better constraint sets, but also give rise to better models.
4.4 Key performance indicators for all targets show that post-selection modeling is consistently better
When summarizing those individual observations for all other targets, we understood that in order to probe the quality improvement conferred by constraint selection, there are three key performance indicators of interest, which are somewhat related: an increase on the proportion of true-positive contacts, the enrichment of the output model set with candidates of correct topology, and the ability to elect a better representative model (or ensemble of models) from such output set. We built Figure 3 to represent the global results based on those elected figures of merit.
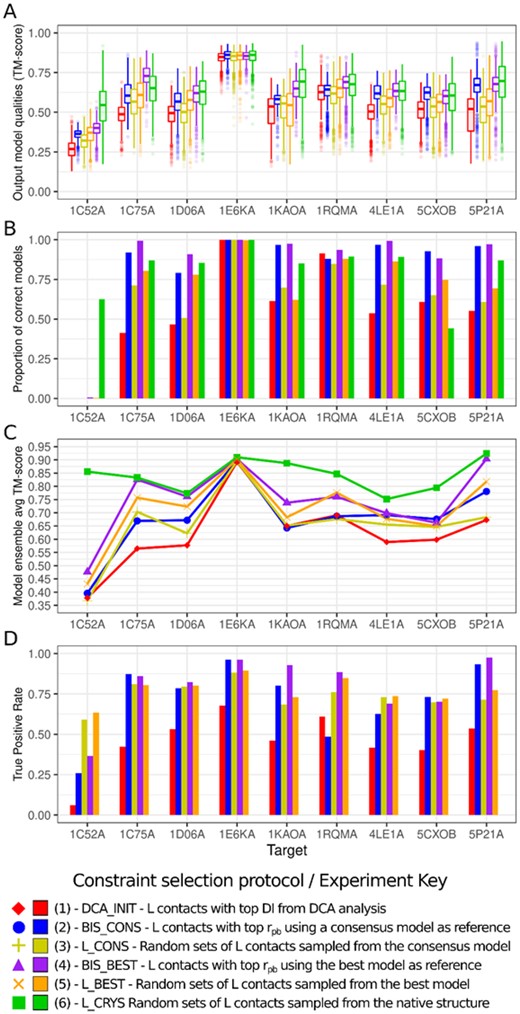
Assessment of key performance indicators of constraint selection results for all nine targets. (A) Boxplots of TM-score of alignment with crystallographic structure, for each target and each Constraint set/Experiment. (B) Proportion of models whose TM-score (in A) was higher than 0.5, for each target and each Constraint set/Experiment. (C) Average TM-score of a blindly elected ensemble of 10 models extracted from the final modeling round in each experiment. (D) True Positive Rate or Percentage of correct contacts in each constraint set, excluding L_CRYS since it contains, by definition, 100% true-positive contacts. For the combinatorial/random sets (L_CONS, L_BEST), reported value is an average over all individual sets generated
In Figure 3A, we address the criterion of overall model topologies by presenting boxplots of model qualities measured as TM-score of the alignment between each model and the reference PDB structure for all targets. There is a notable pattern for the shifting of the mean model quality, which for most cases, is minimal on the preliminary DCA_INIT modeling and reaches a maximum on the idealized L_CRYS set. Notably, the mean model quality was higher after constraint selection (BIS sets) in comparison with the preliminary DCA constraints, advocating the success of constraint selection using point-biserial correlations. This success can be further examined on Figure 3B, which shows the proportion of models generated in the final round in each experiment whose TM-score is greater than 0.5 against the crystallographic structure. With the exception of targets 1C52 (which produced no successful models neither before nor after constraint selection), 1E6K (which showed no significant increase in quality) and 1RQM (which actually experienced a marginal decrease in proportion of successful models), all the other targets underwent a noticeable increase on the amount of correct models, ranging from 50% to 220% increments.
At this moment, it seemed logical to assess whether our proposed use of a consensus model through the point-biserial coefficient was better than simply attempting to populate the neighborhood of this elected model. From Figure 3A, it can be noted that the mean quality is consistently higher on the BIS_BEST and BIS_CONS experiments when compared to their counterparts L_BEST and L_CONS. Figure 3B also confirms that the proportion of models with correct topology follows the same pattern, with the exception of target 1C52. This observation allows us to remark that the employment of a consensus model through our proposed selection mechanism is consistently better performant than the naive use of constraints derived from the consensus model.
Figure 3C portrays the quality of the representative model ensemble, elected via consensus, while Figure 3D represents the proportion of true positives (TPR) on each constraint set. Through examination of the elected representative models, we again notice a consistent pattern where the models generated and blindly elected after point-biserial selection are better than their DCA and naive counterparts, even if marginally.
4.5 Point-biserial contact maps surpass other sets with more true positives due to the lack of trivial constraints
A question that naturally arises, at this point, is how can the BIS_BEST and BIS_CONS constraint sets surpass the naive L_CONS and L_BEST constraint set in model qualities, if the latter portray higher true-positive rates? Or, somewhat equivalent, how can the BIS sets generate a higher proportion of successful models when compared to the L_CRYS set, whose true positive rate is a perfect 100%?
The answer to both questions lies in the relative population of trivial constraints on the combinatorial sets. Although the BIS sets contain fewer true-positives, they are structurally discriminating in such a way that their capability of biasing the conformational space surpasses that of their counterparts, richer in true positives, but rather non-informative ones.
To illustrate this, we produced Figure 4 which summarizes the mean amount of structural information (Censoni and Martínez, 2018) encoded by the constraint set (Fig. 4A). This method measures information by comparing distances in the target structure with their estimated likelihoods, derived by a self-avoiding random walk model of the peptide chain. The most informative constraints are the ‘unexpected’ ones, which might be short-range contacts between distant residues in the sequence, or long-range contacts in general, but particularly those which require stretched conformations of the peptide chain. We also decomposed the true-positive rates reported in Figure 3D in two separate contributions: that accounting only the medium and long-range contacts (Fig. 4B) and that accounting only the short-range contacts (Fig. 4C).
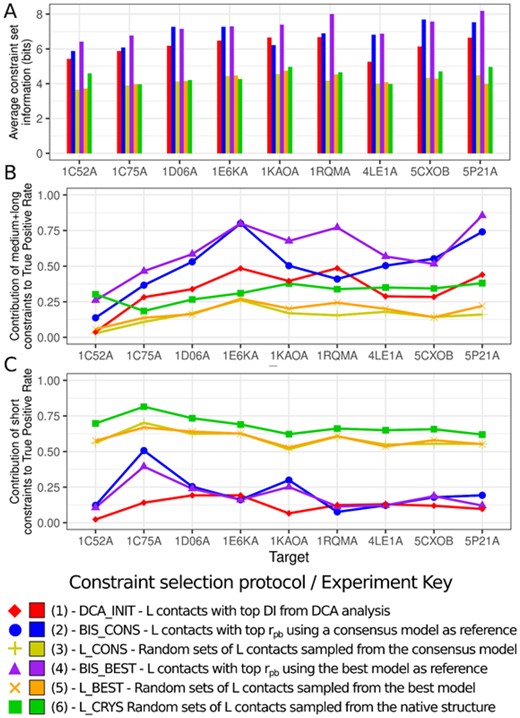
Some additional figures of merit for analysis of the constraint sets. (A) Average structural information (Censoni and Martínez, 2018) contained in each set. (B) Contribution of medium and long-range contacts to the True Positive Rate of the constraint set. (C) Contribution of short-range contacts to the True Positive Rate of the constraint set. Matching points in B and C, combined, reform the TPR reported in Figure 3D. Again, reported values are averages over all individual sets generated in combinatorial experiments
Combined inspection of Figures 3D, 4B and C show an interesting pattern: even though the BIS sets may not have always the highest true-positive rates, their TPR is consistently concentrated on medium and long-range contacts, which are believed to be much more contributing to modeling efforts than shorter constraints (Mandalaparthy et al., 2018). The pattern is clear: most of the true-positive rate of the BIS_BEST and BIS_CONS sets is contributed by medium and long-range contacts, while in the case of the L_CONS, L_BEST and L_CRYS sets, the opposite happens: the TPR is greatly concentrated on short-range contacts. This information is reinforced by Figure 4A, which shows that the information is generally highest in the BIS sets, adding to the idea that the proposed constraint selection strategy has the potential to naturally—meaning, without the necessity of an arbitrary cutoff—filter out short-range contacts and privilege those of larger primary separation.
It is even possible that, in the BIS sets, some marginally violating false-positives may be useful during the modeling cycles, as means to bring the candidate model closer to the neighborhood of the native state on early stages of the conformational search and up until a point where a larger set of collaborating true positives gains larger weight on the energy scores.
In fact, this very statement can explain the reason why, in Figure 3B, the BIS sets (which all contain some percentage of false-positives) sometimes lead to similar or even higher proportions of models with correct topologies than the L_CRYS set. Again, the 100% true-positive L_CRYS constraint sets contain fairly larger amounts of trivial constraints than their BIS counterparts. Because of that, the ability of the L_CRYS constraint sets to effectively shift the mean modeling quality is diminished. In this case, however, since every single constraint is a crystallographic true positive contributing (in a higher or lower magnitude) to the same region of the conformational space, a huge part of the models is concentrated on a fairly small portion of the folding funnel, allowing for the selected consensus model to be of much better topology.
5 Conclusion
Selecting a good subset of constraints from an estimated contact map is critical to the performance of assisted protein structural modeling. In order to respect the particularities of the modeling workflow, a selection criterion should be able to penalize trivial constraints taking some kind of structural measure in consideration. Our proposed indicator, point-biserial correlation coefficient employing consensus similarity as an individual model score, was able to consistently improve modeling experiments starting from a simple coevolution analysis. In the end of three iterations, the constraint sets exhibited a larger amount of true-positives, concentrated on medium and long-range primary separations, leading to an overall increase in the average structural information encoded.
This improvement reflected on the output models as higher average model qualities, higher proportion of correct topologies and a better representative blindly selected model. Significant improvements were found on seven out of nine targets, with marginal but still positive results for the other two. We believe this advocates to the use of constraint selection strategies on different workflows, especially point-biserial correlation coefficient, which, due to the simple principle of being based on structural discrimination, should be able to be modularly inserted on a vast amount of different assisted modeling strategies.
Funding
This work was supported by São Paulo Research Foundation (FAPESP) [2010/16947-9, 2013/08293-7, 2016/13195-2, 2018/14274-9, 2018/24293-0, 2019/17007-4]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [140317/2019-8]; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) [finance code 001].
Conflict of Interest: none declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}