





Abstract
Whole genome sequencing of patient populations is identifying thousands of new variants in untranslated regions (UTRs). While the consequences of UTR mutations are not as easily predicted from primary sequence as coding mutations are, there are some known features of UTRs that modulate their function. utr.annotation is an R package that can be used to annotate potential deleterious variants in the UTR regions for both human and mouse species. Given a CSV or VCF format variant file, utr.annotation provides information of each variant on whether and how it alters known translational regulators including upstream open reading frames, upstream Kozak sequences, polyA signals, Kozak sequences at the annotated translation start site, start codons and stop codons, conservation scores in the variant position, and whether and how it changes ribosome loading based on a model derived from empirical data.
utr.annotation is freely available on Bitbucket (https://bitbucket.org/jdlabteam/utr.annotation/src/master/) and CRAN (https://cran.r-project.org/web/packages/utr.annotation/index.html).
Supplementary data are available at Bioinformatics online.
1 Introduction
Post-transcriptional regulation is essential to the control of translation, controlling the abundance, timing, and location of new protein production. For example, key regulatory elements in untranslated regions (UTRs), such as upstream open reading frames (uORFs) (Barbosa et al., 2013; Calvo et al., 2009), Kozak sequences (Acevedo et al., 2018; Kozak, 1986) and polyA signals (Rehfeld et al., 2013; Weill et al., 2012) strongly influence protein production. Variants that alter these regulatory elements could substantially modify the final protein levels and potentially lead to disease. Thus, annotation of genomic variants in UTRs is essential to reveal potential etiopathogenetic mechanisms. Given the tremendous number of sequence variants being discovered, highlighting putative deleterious variants would be useful to prioritize functional studies, or weight human genetic analyses. Combined Annotation-Dependent Depletion (CADD) (Rentzsch et al., 2019) is widely used to measure variant deleteriousness, however, its algorithm is currently unaware of many key regulatory features of UTRs. A recently developed UTR annotator (Zhang et al., 2020) only annotates uORFs perturbation in 5′ UTR. Therefore, we developed an R package, utr.annotation, that annotates genetic variants’ effect on additional categories of UTR key regulatory elements in both 5′ and 3′ UTRs.
2 An R package for annotating potential deleterious variants in non-coding regions
The utr.annotation package (Fig. 1A) can be used to highlight potentially deleterious variants by annotating its impact on known UTR regulators easily predicted from sequence: uAUGs, upstream Kozak sequences (uKozaks), polyA signals, the Kozak sequence at the annotated translation start site, as well as start codons, and stop codons. In addition, as highly conserved regions tend to have important biological functions, the package can annotate Phastcons and PhyloP conservation scores at the variant positions if the user downloads the conservation track files in bigWig format from UCSC genome browser into a folder and provides the tool with the path to the folder. Finally, the utr.annotation package also leverages a deep learning model trained from empirical data (Sample et al., 2019) using their ‘generalized Optimus 5-prime’ neural network to predict changes in mean ribosome loading (MRL) based on 5′ UTR on reference and alternative alleles. The package has been tested and is suitable for annotating any SNV and short InDels.
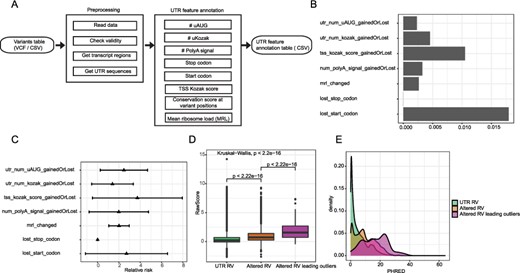
utr.annotation pipeline and its application on identifying high-risk variants of eOutliers in GTEx. A) utr.annotation pipeline. B) Proportion of utr.annotation identified variants that were carried by eOutliers individuals. C) Relative risk of seven variant types. D) CADD raw scores comparison among three groups of variants. E) PHRED distributions of three variant groups.
2.1 Input and output formats
The runUTRAnnotation function is used to annotate any SNV and short InDels for potential UTR regulatory impact. As input, the user provides variant information in either the standard VCF or CSV format including four columns: Chr, Pos, Ref and Alt. They also specify species, a reference genome build and output directory. To increase speed, the user may optionally include a Transcript column for a CSV variant file as well, providing an ENSEMBL format transcript ID overlapping each variant, if already available. The annotation table is outputted in CSV format. The sample test data with input and output are provided in the Supplementary Material. Detailed explanation on the key terms and annotation results are in the Supplementary Tables S1 and S2.
2.2 Robust on genome annotations
The utr.annotation package works robustly on mouse and human variants regardless of genome build. The runUTRAnnotation function will use the latest Ensembl genome annotation by default and you can choose any available Ensembl version for identifying gene/transcript sequences.
2.3 Efficient on large data
For <10 000 variants, running the annotation on a typical PC is feasible and benchmarks at <210 min with 1 CPU. For larger datasets, the tool can run in parallel on multiple cores (e.g. cluster) (Supplementary Fig. S1). Besides leveraging internal parallelism, a large variant file can be partitioned into a user-specified number of small files using the partitionVariantFile function, and partitioned outputs can be concatenated with the concatenateAnnotationResult function. Using these tools, the Genotype-Tissue Expression (GTEx) example below (∼23 million variants) took 54 h when partitioned into 5000 small files and using 4 CPUs for each file.
3 Application on identifying high-risk variants of eOutliers in GTEx
Rare variants (RVs) with large effects tend to contribute to complex disease risk (Gibson, 2012). However, identifying functional RVs in the non-coding regions remains challenging. Many non-coding variant mutations are thought to influence gene expression, and UTR mutations in particular may influence transcript stability either directly (e.g. impacting miRNA binding sites) or indirectly (e.g. influencing the transcript initiation, as translation can either increase or decrease transcript stability). Either circumstance suggests the hypothesis that rare UTR mutations could contribute to unusual transcript levels in a given individual. Therefore, we annotated UTR RVs (allele frequency < 1%) (uRV), and to identify the frequency with which these were associated with outliers in expression across a population of individuals (eOutliers) (Ferraro et al., 2020). The dataset comprised 20 252 genes from 838 samples of transcriptome data in the GTEx project version 8 (v8). We used utr.annotation to annotate all RV from GTEx on genes in the eOutlier data and identified 59 986 uRV altering UTR regulatory elements (num_polyA_signal_gainedOrLost: 6470, utr_num_uAUG_gainedOrLost: 7782, utr_num_kozak_gainedOrLost: 1536, tss_kozak_score_gainedOrLost: 1904, lost_start_codon: 724, lost_stop_codon: 414, mrl_changed: 41 156).
We next compared our annotation to CADD, a standard variant annotation tool. CADD combines numerous different variant features into a single score. It is aware of conservation scores in UTR features, but not ORF, Kozak, PolyA and MRL. We compared the CADD scores of three groups of RVs: uRVs, uRVs that altered UTR feature elements and uRVs that altered UTR feature elements and were carried in eOutliers. uRVs leading eOutliers have significantly higher CADD scores than the other two groups, indicating their higher deleteriousness (Fig. 1D). Specifically, 45 unique uRVs carried in eOutliers had PHRED scores larger than 20, which indicated they are ranked as 1% most deleterious variants in humans (Supplementary Table S3), suggesting some UTR information is being annotated by CADD. However, our annotation also captured 55 potential deleterious variants that were carried in eOutliers but had CADD scores <10 (Supplementary Table S4), suggesting CADD is missing some information. Four of the CADD underestimated variants altered ployA signals in 3′ UTR and two out of four have moderate conservation (phastCons = 0.653 and 0.351). One CADD underestimated variant changed MRL and was in a highly conserved position (phastCons = 0.994). One CADD underestimated variant altered uORF and has high conservation score (phastcons = 1). Overall, our utr.annotation could detect additional presumably harmful variants that are overlooked by current annotation tools, and thus might make a useful addition to the next version of CADD.
Finally, we had note that there are many mechanisms by which a variant might be highly detrimental to protein production, but not influence the transcript levels (the only readout available currently for GTEx). Thus, this annotation tool may show even higher overlap in future studies of protein level outliers as well.
4 Conclusion
Our utr.annotation package is a user-friendly and scalable tool to annotate variants that alter the UTR regulatory elements. It can be used to select top deleterious variants for functional analysis and provide detailed information on UTR interruption for further exploration, or statistical analysis.
Acknowledgements
We would like to thank Paul Sample and the Seelig lab for providing a Kozak PWM and the MRL algorithm, and the Battle lab for the eOutlier lists. We would also like to thank Sergej Djuranovic, Tomas Lagunas, Tony Fischer, Stephen Plassmeyer, Stu Fass, Stephan Sanders and Michael Wells for helpful comments and discussion.
Funding
This work was supported by the Simons Foundation (571009) and the National Institutes of Health (5R01MH116999, 5R01NS10227, R01GM112824, 5R01GM112824-06).
Conflict of Interest: none declared.


{kind=link}