





Abstract
VoroContacts is a versatile tool for computing and analyzing contact surface areas (CSAs) and solvent accessible surface areas (SASAs) for three-dimensional (3D) structures of proteins, nucleic acids and their complexes at the atomic resolution. CSAs and SASAs are derived using Voronoi tessellation of 3D structure, represented as a collection of atomic balls. VoroContacts web server features a highly configurable query interface, which enables on-the-fly analysis of contacts for selected set of atoms and allows filtering interatomic contacts by their type, surface areas, distance between contacting atoms and sequence separation between contacting residues. The VoroContacts functionality is also implemented as part of the standalone Voronota package, enabling batch processing.
Supplementary data are available at Bioinformatics online.
1 Introduction
The knowledge of interatomic interactions and of interactions with solvent is critical for understanding the formation of macromolecular structures and their binding to other macromolecules and ligands. Not surprisingly, over the years multiple approaches have been developed for the analysis of contacts within and between the macromolecular structures, surface burial or exposure to solvent. Solvent accessible surface areas (SASAs) are often computed using algorithms pioneered in the seventies of the last century or their modifications (Lee and Richards, 1971; Mitternacht, 2016; Ribeiro et al., 2019; Shrake and Rupley, 1973). Interatomic contacts can be defined in many different ways, for example, by using distance and (optionally) angle criteria (e.g. Jubb et al., 2017; Scheurer et al., 2018), by applying a rolling probe (e.g. Ribeiro et al., 2019; Word et al., 1999) or by employing structure tessellation methods (e.g. Mach and Koehl, 2013; McConkey et al., 2002; Olechnovič and Venclovas, 2014b). Because of their simplicity distance-based methods are most common. However, the drawback of such methods is that they do not account for structural neighbors which may affect a given interaction (Esque et al., 2011). Also, in that case, the representation of intramolecular contacts and contacts with solvent (SASA) is conceptually different. A more consistent way is to describe both types of contacts through contact surface area (CSA). Such approaches have been shown to be highly effective in quantifying differences between conformations of protein and RNA structures (Olechnovič et al., 2013; Olechnovič and Venclovas, 2014a), comparing interfaces of biological assemblies (Dapkūnas et al., 2017) and developing statistical potentials (McConkey et al., 2003; Olechnovič and Venclovas, 2017). To derive CSAs, some methods use modified SASA computation algorithms (e.g. Ribeiro et al., 2019), others employ structure tessellation approaches (Mach and Koehl, 2013; McConkey et al., 2002). Voronoi diagram of balls, also known as additively weighted Voronoi diagram (Goede et al., 1997), is arguably the best-fitting representation of macromolecular structures for the analysis of interatomic interactions as it unequivocally identifies neighbors and takes into account differences in atom size.
Here, we present VoroContacts, a versatile tool for computation and analysis of interatomic CSAs and SASAs using Voronoi structure tessellation. VoroContacts enables highly configurable contact queries ranging from individual atoms and residues to macromolecular structures and their assemblies. In addition, contacts can be filtered by different properties, interactively analyzed and visualized.
2 Implementation
For computation of CSA and SASA, macromolecular structure is represented as the Voronoi diagram of atomic balls using Voronota (Olechnovič and Venclovas, 2014b). Balls correspond to heavy atoms of specified van der Waals (VDW) radii. For proteins VoroContacts uses the set of VDW radii compiled by Li and Nussinov (1998). All the radii values used for proteins, other macromolecules, ligands and ions are listed in Supplementary Tables S1–S3. Contacts and their surface areas are defined as described previously (Olechnovič and Venclovas, 2017). Briefly, a geometric representation of CSA between two atoms corresponds to a boundary surface (Voronoi face) shared between Voronoi cells of these atoms. In general, the surface is non-planar as different atom types have different radii. To compute SASA, an intersection of a Voronoi cell and an atomic sphere having a VDW radius extended by the radius of water molecule (1.4 Å) is considered. If the faces of the Voronoi cell are all enclosed within the extended atomic sphere, the atom is completely buried (surrounded by neighbors). If any of the faces of the Voronoi cell extend beyond the sphere, CSAs correspond to surfaces of the faces constrained by the sphere, whereas the SASA corresponds to the surface patch of the sphere cut out by the Voronoi cell. The SASA of an atom may be considered as the upper limit of the sum of CSAs corresponding to all possible positions of solvent atoms in contact with the atom of a macromolecule. Both Voronoi faces (interatomic CSAs) and spherical surfaces (SASAs) are represented by triangulation, which is used both to visualize and to quantify CSAs and SASAs. Importantly, the Voronoi tessellation procedure naturally takes into account the structural context, that is, the extent neighboring atoms have a shielding effect on a pair of interacting atoms. For a set of atoms such as a residue, an individual macromolecular structure or a complex, CSAs and SASAs are computed by simply summing the corresponding values of the constituent atoms. Figure 1 illustrates the construction of interatomic contact surfaces and their integration for residues and protein structures. In addition to surface areas, interatomic contacts are annotated by the distance between the centers of contacting atoms and whether the contact represents a hydrogen bond (hb), a salt bridge (sb) or a disulfide bridge (ds). hbs are identified with HBPLUS (McDonald and Thornton, 1994).
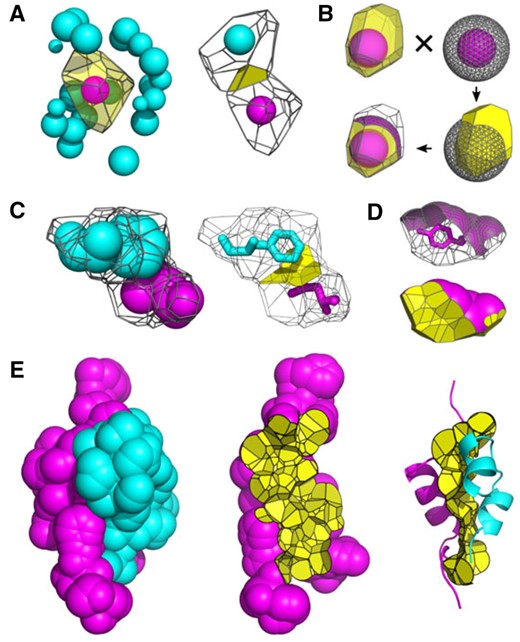
Illustration of contact surfaces at different scales. (A) Atom–atom contacts: (left) Voronoi cell of an atom surrounded by its neighbors and (right) atom–atom CSA defined as the face shared by two adjacent Voronoi cells. (B) Atom–solvent contacts: the SASA of an atom corresponds to the surface patch of the extended sphere cut out by the Voronoi cell. (C) Residue–residue contacts: (left) Voronoi cells of two neighboring residues and (right) residue–residue CSA defined as a union of atom–atom CSAs. (D) Residue–solvent contacts: the SASA of a residue is the union of atomic SASAs. (E) SASAs and intersubunit interfaces: (left) the SASA of an insulin heterodimer (PDB: 4ung) colored by subunit, (center) the intersubunit interface shown together with the SASA of one subunit and (right) the intersubunit interface shown together with both subunits represented as cartoons
The performance of VoroContacts was assessed by comparing it with a recently published method, dr_sasa (Ribeiro et al., 2019), which is also capable of computing both SASAs and interatomic CSAs. Comparison revealed that the agreement between the tools in calculating per-atom SASA is nearly perfect. Values for atom–atom and residue–residue CSAs were also strongly correlated (Pearson correlation coefficients of 0.83 and 0.97, correspondingly), but dr_sasa showed a tendency of reporting larger CSAs than those computed by VoroContacts. Additionally, the impact of the choice of rolling probe radius was investigated. Again, both methods showed essentially identical sensitivity in computing SASAs, but VoroContacts was significantly less sensitive in computing CSAs. Thus, it can be concluded that dr_sasa and VoroContacts agree well on SASAs, but show some differences in computing CSAs. These differences may be explained by the fact that the Voronoi tessellation, used by VoroContacts, takes into account not only the proximity of atoms to each other, but also their structural context, which may have the shielding effect on their contact. Tests revealed that VoroContacts is not only more robust, but also significantly faster than dr_sasa. Detailed results of the comparison are presented in Supplementary Figures S1–S7.
3 Usage
The VoroContacts server accepts structures of biological macromolecules (proteins, RNA, DNA) and their complexes. The structure may be uploaded as a local file in either PDB or mmCIF format. Alternatively, the structure may be fetched directly from PDB. Once the job is submitted, CSAs and SASAs are computed for individual atoms of the entire structure and the results are prepared for interactive analysis using a form for querying contacts between the two user-defined sets of atoms. These two sets can be defined at the level of atoms, residues and chains in various combinations. SASA is defined as the contacts between the structure atoms and the solvent, assigned a special chain name ‘solvent’. In addition, contacts themselves can be filtered by several criteria, including surface area size, distance between atoms, sequence separation between contacting residues and by the contact type (hb, ds or sb). Query results consist of a summary of the matched contacts and the table providing a detailed list of individual contacts and their attributes. The results table is color-coded in the same way as the query form in order to make easy connection between the two. Information on contacts can be downloaded as a text file for local analysis. The script to reproduce the query results offline using the Voronota package is also available for downloading. CSAs/SASAs can also be visualized online or can be downloaded as a PyMol graphics object wrapped in a script.
In summary, VoroContacts and the standalone Voronota package implementing the corresponding functionality enable users to ask a large variety of questions related to interactions within and between biological macromolecules as well as their interactions with ligands and solvent. Typical applications might include calculation of SASA and buried surface area, detection and annotation of interactions in protein–protein, protein–DNA/RNA or protein–ligand complexes. In addition to these standard applications, many more customized analyses of interactions at various levels of detail (atom, residue, substructure or subunit) can be performed.
Acknowledgements
The authors wish to thank Saulius Gražulis, Giedrė Tamulaitienė and Justas Dapkūnas for useful comments and suggestions.
Funding
This work was supported by the European Social Fund [09.3.3-LMT-K-712-01-0080] under grant agreement with the Research Council of Lithuania (LMTLT).
Conflict of Interest: none declared.


{kind=link}