





ABSTRACT
Currently, most computational methods estimate the expression of circular RNAs (circRNAs) using the number of sequencing reads that support back–splicing junctions (BSJ) in RNA-seq data, which may introduce biased estimation of circRNA expression due to the uneven distribution of sequencing reads. To overcome this, we previously developed a model-based strategy for circRNA quantification, enabling consideration of sequencing reads from the entire transcript. Yet, the lack of exact transcript structure of circRNAs may limit its accuracy. Here, we proposed a substantially improved circRNA quantification tool, AQUARIUM, by introducing the full-length RNA structure of circular isoforms. We assessed its performance in circRNA quantification using both biological and simulated rRNA-depleted RNA-seq datasets, and demonstrated its superior performance at both BSJ and isoform level.
AQUARIUM is freely available at https://github.com/wanjun-group-seu/AQUARIUM.
Supplementary data are available at Bioinformatics online.
1 Introduction
Circular RNAs (circRNAs) are a group of non-canonical RNAs with important biological functions (Chen, 2020), and their aberrant expression is related to many diseases (Wen et al., 2020). Thus, the precise estimation of circRNA expression from ribosomal RNA (rRNA)-depleted RNA-seq data is crucial in circRNA-related biomedical research (Gao and Zhao, 2018). Recent studies have developed several computational tools to estimate the abundance of circRNAs, including CIRI2 (Gao et al., 2018), KNIFE (Szabo et al., 2015), CIRCexplorer2 (Zhang et al., 2016), CLEAR (Ma et al., 2019), CircAST (Wu et al., 2020), seekCRIT (Chaabane et al., 2020) and CIRIquant (Zhang et al., 2020). All these tools quantified circRNA expression by counting the number of sequencing reads that support back–splicing junctions (BSJ). However, sequencing reads are not evenly distributed along with both linear and circular transcripts in high-throughput sequencing (Wu et al., 2011). Therefore, using the number of sequencing reads that cross BSJ sites to represent the expression of circular transcripts may cause biased estimation of circRNA expression, which would be even more concerned, given that most circRNAs are expressed at relatively low levels (Guo et al., 2014). In our previous study, we proposed a model-based quantification strategy, Sailfish-cir, to quantify circRNA expression (Li et al., 2017). Sailfish-cir transforms circular transcripts to pseudo-linear transcripts and quantifies circRNA expression using sequencing reads that are originated from the entire transcript, rather than BSJ only (Li et al., 2017). The estimation accuracy of Sailfish-cir is quite dependent on the exact structure of circRNA transcript. However, it is largely unknown regarding the full-length sequence information of most circRNAs identified by existing identification tools, such as CIRI (Gao et al., 2015), which may, to some extent, limit the performance of Sailfish-cir (Zhang et al., 2020). In three recent studies, Zheng et al. (2019), Xin et al. (2021) and Zhang et al. (2021) have investigated the transcript structure of circRNAs and partially addressed the problem of full-length circRNA reconstruction. Zheng et al. (2019) presented a computational approach, CIRI-full, for effective reconstruction of full-length circRNAs from short-read RNA-seq dataset. Instead, Xin et al. (2021) and Zhang et al. (2021) experimentally determined the full-length sequences of circRNAs using rolling circle amplification and Nanopore long-read sequencing strategy. Based on these advances, we proposed a state-of-the-art model-based circRNA quantification pipeline, AQUARIUM (Accurate QUAntification of circulaR Isoforms Using Model-based strategy), by introducing full-length sequences and exact exonic structures of circRNA isoforms. We evaluated the performance of AQUARIUM on circRNA quantification at the BSJ and isoform level in both biological and simulated RNA-seq datasets. Our results demonstrated that AUQARIUM can substantially improve the accuracy of circRNA quantification and deliver better performance compared with the existing count-based tools.
2 Overview
2.1 Aquarium procedures
Unlike Sailfish-cir (Li et al., 2017), AQUARIUM integrated sequence and exonic structure of full-length circRNA isoforms from CIRI-full output (Zheng et al., 2019), rather than the BSJ information from CIRI output (Gao et al., 2015), to quantify the expression of both circular and linear transcripts from rRNA-depleted RNA-seq datasets (Fig. 1A). First, a set of circRNA reference sequences was compiled using CIRI-full circRNA identification and reconstruction. For the circRNA isoforms that are identified as the full-length transcript, the complete reconstructed sequences from CIRI-full output were used as the reference templates. For the circRNA isoforms that are not completely characterized by CIRI-full, the reconstructed partial sequences from CIRI-full output and all the exonic sequences in the un-reconstructed regions were joined as the reference sequences. For the circRNAs with BSJ information only, all the known exonic sequences between the BSJ sites were concatenated as the putative references. Next, circular transcripts were transformed to pseudo-linear transcripts as we performed in Sailfish-cir (Li et al., 2017). Then, the transformed pseudo-linear reference sequences and all the known linear RNA reference sequences from genomic annotation were merged together as the reference transcripts. Finally, Salmon (Patro et al., 2017) was used to simultaneously quantify the expression of both circular and linear transcripts from the RNA-seq data.
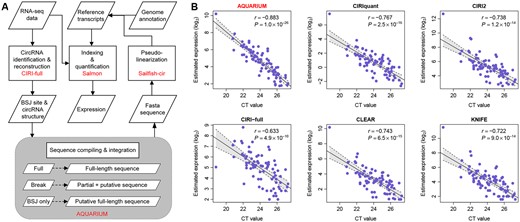
(A) The AQUARIUM workflow to estimate expression values of both circular isoforms and linear RNA transcripts from rRNA-depleted RNA-seq data. (B) Comparison of the quantification performance between the AQUARIUM, CIRIquant, CIRI2, CIRI-full, CLEAR and KNIFE. X-axis and Y-axis represent the circRNA expression measured by RT-qPCR and the estimated circRNA expression by each tool from RNA-seq data (log2-transformed), respectively. The expression data were recorded as TPM for AQUARIUM, CPM for CIRIquant, FPBcirc for CLEAR and number of BSJ reads for CIRI2, CIRI-full and KNIFE. The r and P were computed by Pearson correlation test between X- and Y-axes
2.2 Applications
We evaluated the performance of AQUARIUM in both the biological and simulated rRNA-depleted RNA-seq datasets (Supplementary Methods). In the biological RNA-seq dataset, the estimation of AQUARIUM was highly correlated with the expression values experimentally determined by RT-qPCR (n = 78; r = −0.883 and P = 1.0 × 10−26) in 11 human fetal tissues at different developmental time points (Szabo et al., 2015) (Fig. 1B). More encouragingly, in comparison to the existing computational tools that quantify circRNA expression at the BSJ level, i.e. CIRIquant, CIRI2, CIRI-full, CLEAR and KNIFE, the AQUARIUM estimates exhibited the highest consistency with the RT-qPCR readouts (Fig. 1B), while the count-based tools had the relatively lower accuracy in circRNA quantification (Fig. 1B and Supplementary Fig. S1). The superior performance of AQUARIUM was observed in the simulated RNA-seq datasets as well (Supplementary Fig. S2). Comparing to Sailfish-cir, AQUARIUM substantially increased its estimation accuracy by using full-length circular transcripts (Supplementary Fig. S3). Furthermore, we found that the concordance between the reference transcripts used in AQUARIUM and the simulated circular transcripts (Supplementary Fig. S3 and Supplementary Fig. S4) is the major factor affecting the estimation accuracy, rather than the sequencing depth (15 M, 30 M and 60 M reads) (Supplementary Fig. S2) and read length (PE100, PE150 and PE250) (Supplementary Fig. S5). This is mainly due to the non-uniform distribution of sequencing reads along with the circular transcripts (Supplementary Fig. S6). In addition to the quantification of circular transcripts at the BSJ level, we also examined the performance of AQUARIUM in estimating circRNA expressions at the isoform level. Using an rRNA-depleted RNA-seq data of human Hela cell line (Zheng et al., 2019), we found that the expression of the circRNA isoforms quantified by AQUARIUM was significantly correlated with the RT-qPCR readouts (n = 12; r = −0.828 and P = 8.9 × 10−4), which outperformed the CIRI-full tool (Supplementary Fig. S7A). The similar results were observed in the simulated RNA-seq data, in which 109 alternatively spliced circular isoforms were included along with AQUARIUM showing a superior quantification accuracy compared with CIRI-full (Supplementary Fig. S7B).
2.3 Availability and implementation
AQUARIUM is implemented using Python scripts, which is freely accessible at https://github.com/wanjun-group-seu/AQUARIUM. The scripts to generate the simulation data, along with the simulated RNA-seq datasets were also available at https://github.com/wanjun-group-seu/AQUARIUM.
3 Conclusion
We proposed a state-of-the-art computational tool for circRNA quantification by integrating full-length sequence and isoform structure of circular transcripts. The inclusion of full-length circular isoforms substantially increases the estimation accuracy of circRNA expression in our model-based framework, which ensures better performance against the existing count-based algorithms. With more full-length circular isoforms identified by computational algorithms (Zheng et al., 2019) or experimental investigations (Xin et al., 2021; Zhang et al., 2021), AQUARIUM will greatly improve the accuracy of circRNA quantification from RNA-seq data.
Acknowledgements
The authors thank Dr Fangqing Zhao for kindly sharing the RT-qPCR data in CIRIquant study.
Funding
This work was funded by grants from the National Key R&D Program of China (2018YFC1314900, 2018YFC1314902 to W.G.), National Natural Science Foundation of China (61571109 to W.G.) and the Fundamental Research Funds for the Central Universities (2242017K3DN04 to W.G.).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, the Guoxia Wen and Musheng Li should be regarded as Joint First Authors.


{kind=link}