





Abstract
fastsimcoal2 extends fastsimcoal, a continuous time coalescent-based genetic simulation program, by enabling the estimation of demographic parameters under very complex scenarios from the site frequency spectrum under a maximum-likelihood framework.
Other improvements include multi-threading, handling of population inbreeding, extended input file syntax facilitating the description of complex demographic scenarios, and more efficient simulations of sparsely structured populations and of large chromosomes.
fastsimcoal2 is freely available on http://cmpg.unibe.ch/software/fastsimcoal2/. It includes console versions for Linux, Windows and MacOS, additional scripts for the analysis and visualization of simulated and estimated scenarios, as well as a detailed documentation and ready-to-use examples.
1 Introduction
Coalescent theory (Kingman, 1982) has provided a very efficient framework to simulate the diversity of neutrally evolving loci (Hudson, 1990; Kelleher and Lohse, 2020; Marjoram and Wall, 2006). These simulations have been used extensively to check the validity of theoretical derivations, and to make predictions of the effect of complex demographic processes on the genomic diversity of populations. Due to their versatility, they have also been used in Approximate Bayesian Computations (ABC, Beaumont et al., 2002) to estimate parameters under very complex models and to perform model testing (Beaumont, 2019; Currat et al., 2019; Mondal et al., 2019; Sanchez et al., 2020; Wegmann et al., 2010). Several faster alternatives to ABC have been developed in the last ten years to estimate demographic parameters under relatively complex scenarios (Albers and McVean, 2020; Gutenkunst et al., 2009; Steinrücken et al., 2019; Weissman and Hallatschek, 2017), many of them fitting the Site Frequency Spectrum (SFS) using exact derivations or approximations (e.g. Excoffier et al., 2013; Gutenkunst et al., 2009; Kamm et al., 2020; Liu and Fu, 2020). It has been shown that the expected SFS could be robustly estimated using coalescent simulations (Excoffier et al., 2013). A clear advantage of SFS-based methods is that the computing time is independent of the length of the analyzed genome. SFS-based methods, however, ignore information on linkage between sites, an information that is used in Hidden Markov Models-based approaches (e.g. Li and Durbin, 2011; Schiffels and Wang, 2020; Speidel et al., 2019; Terhorst et al., 2017) or those based on the Ancestral Recombination Graph (e.g. Gronau et al., 2011; Kelleher et al., 2019). In this paper, we describe the latest implementation of fastsimcoal2, a coalescent-based program that can estimate parameters from SFS under very complex demographic scenarios including continuous arbitrary size changes, gene flow, admixture events, bottlenecks, populations splitting, population growth, inbreeding, serial sampling and spatially structured populations. Compared to its initial release one decade ago (Excoffier and Foll, 2011), fastsimcoal has been extended in several ways described below.
2 Novelties implemented in fastsimcoal2
fastsimcoal became fastsimcoal2 (abbreviated fsc2 in the following) with the implementation of demographic and mutation parameters inference from the SFS (Excoffier et al., 2013). While fsc2 might not have a clear edge over other coalescent simulators of genomic diversity, like e.g. msprime (Kelleher and Lohse, 2020), its innovation is rather in its built-in ability to perform parameter inference under complex evolutionary scenarios, and the most recent developments have therefore focused on this aspect.
2.1 Parameter inference
For parameter inference, coalescent simulations are used to estimate the expected SFS following Nielsen (2000), and a multinomial likelihood (Adams and Hudson, 2004) is maximized using a conditional expectation maximization algorithm (Meng and Rubin, 1993) to estimate the parameters, one at a time over several optimization cycles. This approach has been shown to be very robust (Excoffier et al., 2013) and can, in principle, be applied to an arbitrarily large number of populations, whereas approaches based on analytically derived SFS can only handle a few populations [e.g. 3 populations in ∂a∂i, (Gutenkunst et al., 2009) or 4–5 in dadi. CUDA (Gutenkunst, 2021)], or do not deal with continuous gene flow [e.g. in momi2 (Kamm et al., 2020)]. The trade-off for this robustness and versatility is that computing time, which is independent of genome size, will however increase linearly with the number of sampled genomes, but it remains reasonable given the speed improvements mentioned below. fsc2 also gives the possibility to optimize the likelihood of the model considering all sites (monomorphic and polymorphic), polymorphic positions only or a mixed approach where optimization is first performed using likelihoods based on all sites, and then only considering polymorphic sites after a given number of cycles (-l command line option). Finally, it is now possible to ignore singleton sites (--nosingleton option), which might be useful when considering ancient DNA or low coverage data where some genotyping errors might have arisen. Note that while fsc2 is using the SMC’ approximation (Marjoram and Wall, 2006) of the Sequential Markov Coalescent (McVean and Cardin, 2005) for simulating diversity at linked sites, the SFS estimation is based on the simulation of independent coalescent gene trees.
2.2 Speed improvement
As compared to the first (but unpublished) version of fastsimcoal2 (fsc21), several speed improvements have been performed. First, multi-threading has been introduced using the openMP framework (https://gcc.gnu.org/onlinedocs/libgomp/), allowing one to distribute independent simulations over several threads (-c option). Second, icsilog (Vinyals and Friedland, 2008), a fast approximation of the log function (used to generate exponentially distributed coalescent times) is now used in fsc2, the precision of which can be specified by the user (--logprecision option). Full precision is used by default (–logprecision 23), but computing speed can be improved by 10–25% by slightly lowering the precision (e.g. --logprecision 18)(see Fig. 1B–D). We have also optimized the simulation of large recombining chromosomes, obtaining a > 5× gain for the simulation of 1 Gb-long chromosomes (Fig. 1A). Finally, we have optimized the simulations of samples drawn from large, subdivided populations (e.g. in a 2D stepping-stone), also leading to a drastic speed gain (6×–60×) for such simulations (see Fig. 1C and D).
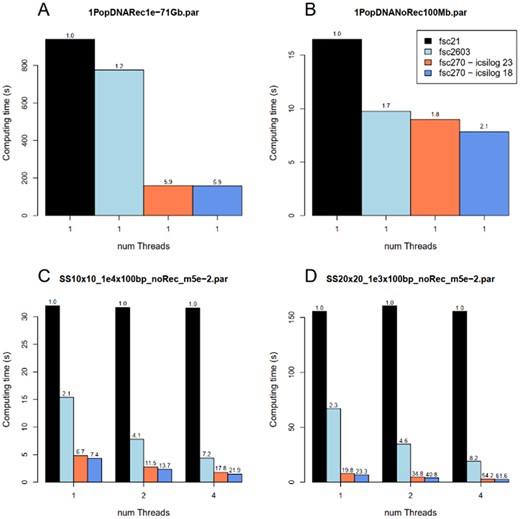
Speed comparison between different versions of fsc2. fsc21: released in 2013, single-threaded. fsc2603: released in 2017, multi-threading but no log acceleration. fsc27: current release, log acceleration, optimized for large chromosomes and highly subdivided populations. (A) Simulation of 100 1 Gb chromosomes, r = 1e-7. (B) Simulation of 100 haploid genomes consisting of 1 million unlinked segments of 100 bp, u = 1.4e-8. (C) Simulation of 2 haploid genomes of 10 000 unlinked segments of 100 bp in a 2D stepping-stone of 10×10 demes. (D) Simulation of 2 haploid genomes of 1000 unlinked segments of 100 bp in a 2D stepping-stone of 20×20 demes. In the two top cases, the mutation rate u = 1.4e-8 per bp, and the haploid population size is 20 000. In the two bottom cases, u = 1.25e-8, m = 0.05 to each of the 4 adjacent demes and the haploid population size of each deme is 200. The numbers above the bars indicate the speed gain factor as compared to fsc21
2.3 Input file syntax enhancement and new command line options
The syntax of input files has been enriched to facilitate the specification of complex evolutionary scenarios. In input parameter (.par) and template (.tpl) files, it is now possible to specify an inbreeding coefficient for each population in the sample section after the sampling times of the simulated lineages, which can be useful when samples are drawn from a subdivided population leading to a Wahlund effect (Wahlund, 2010) or when modeling a true inbred population. One can also define instantaneous bottlenecks (with the instbot keyword) in the historical events section. These bottlenecks are implemented as a single generation of intense rate of coalescence, and their intensity can be specified, where t is the duration of the bottleneck in generations and N is the effective size during the bottleneck. This implementation avoids the need to specify two separate parameters like the population size during the bottleneck N and its duration t, while leading to the same rate of coalescence. Population size changes are now also made simpler, as it is possible to resize a given population size to an absolute value (using the absoluteResize keyword) rather than to a relative value that often used to be computed as a complex parameter.
New options have also been made available in the parameter specification (.est) file. The most useful one is the possibility to specify the search range of a simple parameter as being bounded by the values of previously defined parameters using the paramInRange keyword. For instance, in the following example a bottleneck time (TBOT) can be defined to occur between two divergence times (TDIV1 and TDIV2), with TDIV2 being necessarily larger than TDIV1 as
1 TDIV1 unif 100 10000 output
1 TDIV2 unif TDIV1 10000 output paramInRange
1 TBOTunif TDIV1 TDIV2 output paramInRange
Note that the combination of an absolute value and a parameter for the lower or upper bound can also be provided. Finally, new operations are now possible for the definition of complex parameters: abs(X) for computing the absolute value of X; X%min%Y and X%max%Y for finding the minimum and maximum of the numbers X and Y, respectively; <condition>? <if true>: <if false> for assigning values to a parameter depending on whether a condition is met.
Command line options now include the possibility to define initial parameter values for parameter estimation (--initvalues), which is useful when performing bootstrap confidence interval estimations. Finally, the 1D or 2D folded SFS can be computed in different populations using the --foldedSFS option, which simply folds the unfolded SFS irrespective of what is the overall minor allele among all populations, so as to provide compatibility with ANGSD (Korneliussen et al., 2014) folded SFS.
2.4 Additional tools for the analysis and visualization of the results
Several tools have been developed to facilitate the use of fsc2 and the analysis of the outputs it produces (see http://cmpg.unibe.ch/software/fastsimcoal2/additionalScripts.html). They include a shiny application and several bash, R and python scripts to 9i) prepare input files from VCFs, (ii) resample individuals in genomic blocks of arbitrary size for block bootstrap analyses, (iii) generate parametric bootstrap replicates, (iv) convert multidimensional SFS into a series of 1D and 2D SFS to visually compare observed and expected SFS, (v) identify the least well fitted SFS entries under a given model and (vi) visually inspect an evolutionary scenario embedded in an input (.par) file.
3 Conclusion
fsc2 is a very versatile coalescent simulator able to handle evolutionary scenarios of arbitrary complexity. It can also be used to estimate demographic parameters under similarly complex scenarios from the site frequency spectrum, in a very consistent way. It can now also be used to analyze geographically structured populations in a faster way than some spatially explicit simulators [e.g. SPLATCHE3 (Currat et al., 2019)], even though input files can still be very large as they can require an explicit definition of big migration matrices. The syntax of input file has been improved to build complex scenarios in a simpler and consistent way, eliminating the need of defining rules to establish a hierarchy among parameters. fsc2 is restricted to the simulation of neutral markers, but it can have a wide range of applications from the simulation of whole recombining genomes with complex architectures, to the estimation of parameters in models including many populations exchanging arbitrary and changing numbers of migrants over time, or model comparisons via likelihood-ratio tests or AIC. It has been applied to a variety of organisms including humans (e.g. Malaspinas et al., 2016; Pouyet et al., 2018), animals (Armstrong et al., 2021; de Manuel et al., 2016; Marques et al., 2019, 2018; Meier et al., 2017), plants (González-Martínez et al., 2017; Lu et al., 2019) or microbes (Montano et al., 2015; Vázquez-Rosas-Landa et al., 2020), and it can deal with ancient DNA samples and establish their relationships with modern samples (e.g. Sikora et al., 2019, 2017).
Acknowledgements
The authors thank the following people for their comments on and their testing of fsc2: Isabel Alves, Alexandre Thiéry, Matthieu Foll and Miguel Arenas Busto.
Funding
This work was supported by the Swiss National Science Foundation [310030_188883 to L.E. and 31003A_163338 to L.E. and Ole Seehausen]. V.C.S. was funded by the Portuguese National Science Foundation − Fundação para a Ciência e a Tecnologia [FCT; CEECINST/00032/2018/CP1523/CT0008 and UIDB/00329/2020 granted to cE3c].
Conflict of Interest: none declared.


{kind=link}