





Abstract
A few algorithms have been developed for splitting the genome in nearly independent blocks of linkage disequilibrium. Due to the complexity of this problem, these algorithms rely on heuristics, which makes them suboptimal.
Here, we develop an optimal solution for this problem using dynamic programming.
This is now implemented as function snp_ldsplit as part of R package bigsnpr.
Supplementary data are available at Bioinformatics online.
Introduction
A few algorithms have been developed for splitting the genome in nearly independent blocks of linkage disequilibrium (Berisa and Pickrell, 2016; Kim et al., 2018). Dividing the genome in multiple smaller blocks has many applications. One application is to report signals from independent regions of the genome (Berisa and Pickrell, 2016; Ruderfer et al., 2018; Wen et al., 2017). Another application is for the development of statistical methods, e.g. for deriving polygenic scores (Ge et al., 2019; Mak et al., 2017; Zhou and Zhao, 2020), estimating genetic architecture and performing other statistical genetics analyses (Shi et al., 2016; Wen et al., 2016). Indeed, most statistical methods based on summary statistics also use a correlation matrix (between variants), and these methods often perform computationally expensive operations such as inversion and eigen decomposition of this correlation matrix. These operations are often quadratic, cubic or even exponential with the number of variants. However, if we can decompose the correlation matrix in nearly independent blocks, then we can apply these expensive operations to smaller matrices with less variants, making these operations much faster, and parallelizable. For instance, inverting a block-diagonal matrix requires only inverting each block separately.
Implementation
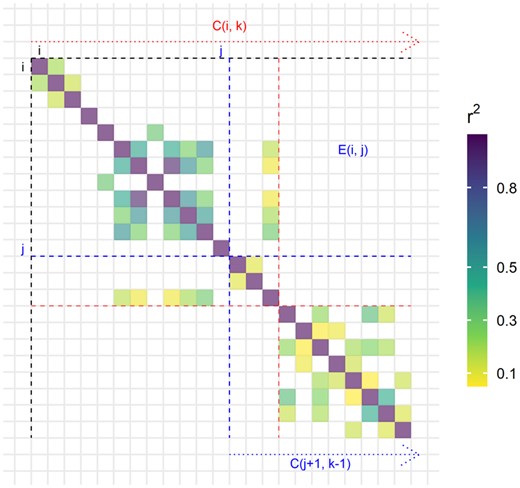
Illustration of subproblems solved by the algorithm using a small LD matrix. The cost of separating the region starting at variant i in k blocks exactly, C(i, k), is broken down in two: the error E(i, j), the sum of all squared correlations between variants from block (i, j) and variants from all the later blocks, and the cost of separating the rest starting at using blocks. The variant j at which the split occurs is chosen so that the cost is minimized. The optimal split is highlighted in red here.
Results
As input, function snp_ldsplit uses a correlation matrix in sparse format from R package Matrix, which can be computed using the available snp_cor function from R package bigsnpr (Privé et al., 2018). This function is fast and parallelized. Then, to run snp_ldsplit using a correlation matrix for 102 451 variants from chromosome 1, it takes <6 min on a laptop to find the optimal split in K blocks (for all K = 1 to 133) with a bounded block size between 500 and 10 000 variants. Then, the user can choose the desired number of blocks, which is a compromise between having more (smaller) blocks with a higher overall cost (LD between blocks), and having less (larger) blocks with a smaller cost. For chromosome 1 and Europeans, ldetect report 133 linkage disequilibrium (LD) blocks (Berisa and Pickrell, 2016); however, we find that they can hardly be considered truly independent given the high cost (10 600) of the corresponding split (Supplementary Fig. S1). When splitting chromosome 1 for Europeans using the optimal algorithm we propose here, it can be split into 39 blocks at a cost of 1, in 65 blocks at a cost of 10, and in 133 blocks at a cost of 296 (Supplementary Fig. S1). Similar results are found for other chromosomes, and for Africans and Asians; however, splitting the LD from admixed Americans comes at a high cost (Supplementary Figs S2–S5). Both methods largely pick block boundaries at recombination hotspots (Supplementary Figs S7 and S8). We also provide an application to LD score regression in Supplementary section ‘Application to LD score regression’, where we show that standard errors for the SNP heritability using nearly independent blocks tend to be larger than when there is substantial LD between blocks, especially for phenotypes with large associations in the HLA (human leukocyte antigen) region (a long-range LD region).
Software, code and data availability
The newest version of R package bigsnpr can be installed from GitHub (see https://github.com/privefl/bigsnpr). All code used for this article is available at https://github.com/privefl/paper-ldsplit/tree/master/code. The HapMap3 variants annotated with 242 blocks can be downloaded at https://www.dropbox.com/s/hdui60p9ohyhvv5/map_blocks.rds?dl=1. LD score regression results are available at https://github.com/privefl/paper-ldsplit/tree/main/ldsc_blocks, with a description of the 245 phenotypes used at https://github.com/privefl/UKBB-PGS/blob/main/phenotype-description.xlsx.
Acknowledgements
We thank Bjarni J. Vilhjálmsson for his feedback on the manuscript, and the two reviewers for their comments and suggestions.
Funding
This work was supported by the Danish National Research Foundation (Niels Bohr Professorship to Prof. John McGrath).
Conflict of Interest: none declared.


{kind=link}