





Abstract
High-throughput experiments are generating ever increasing amounts of various -omics data, so shedding new light on the link between human disorders, their genetic causes and the related impact on protein behavior and structure. While numerous bioinformatics tools now exist that predict which variants in the human exome cause diseases, few tools predict the reasons why they might do so. Yet, understanding the impact of variants at the molecular level is a prerequisite for the rational development of targeted drugs or personalized therapies.
We present the updated MutaFrame webserver, which aims to meet this need. It offers two deleteriousness prediction softwares, DEOGEN2 and SNPMuSiC, and is designed for bioinformaticians and medical researchers who want to gain insights into the origins of monogenic diseases. It contains information at two levels for each human protein: its amino acid sequence and its three-dimensional structure; we used the experimental structures whenever available, and modeled structures otherwise. MutaFrame also includes higher-level information, such as protein essentiality and protein–protein interactions. It has a user-friendly interface for the interpretation of results and a convenient visualization system for protein structures, in which the variant positions introduced by the user and other structural information are shown. In this way, MutaFrame aids our understanding of the pathogenic processes caused by single-site mutations and their molecular and contextual interpretation.
Mutaframe webserver at http://mutaframe.com/.
Supplementary data are available at Bioinformatics online.
Whereas the amount of genetic data obtained through high-throughput sequencing experiments has exploded in the last twenty years (1000 Genomes Project Consortium, 2015), it remains challenging to accurately predict and interpret how some gene variants lead to diseases, which are often caused by changes in the protein(s) the gene encodes (Andreoletti et al., 2019). Especially difficult to predict are the changes these variants cause at the level of protein behavior, which can often explain the pathogenic mechanisms involved and allows optimizing the rational development of targeted drugs. Multiple bioinformatics tools have been developed to classify variants in the human exome as deleterious or neutral (Chen et al., 2020; Livesey and Marsh, 2020), but their explanatory power remains limited.
We present a substantial extension of the Mutaframe webserver (Raimondi et al., 2017), which is designed to improve the interpretability of such protein-level predictions via an easy-to-use graphical interface (Fig. 1). The new version features two complementary state-of-the-art predictors, DEOGEN2 (Raimondi et al., 2016, 2017) and SNPMuSiC (Ancien et al., 2018). DEOGEN2 is a protein sequence-based predictor that utilizes evolutionary information as well as contextual information, such as the relevance of the gene containing the variant or the interactions of the encoded protein. SNPMuSiC uses as input experimental or modeled three-dimensional (3D) protein structures and predicts deleterious variants on the basis of the changes in stability these cause.
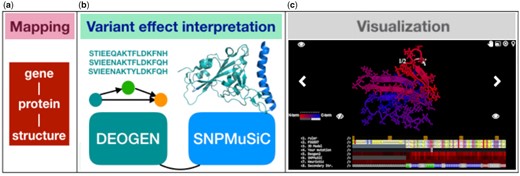
The capabilities of the MutaFrame webserver at the mapping (a), variant effect interpretation (b) and 3D structure visualization (c) levels (Supplementary Material)
Combining these two predictors, which already individually have good performances (Chen et al., 2020; Livesey and Marsh, 2020), yields a consensus predictor with a balanced accuracy of 92% and a positive predictive value of 97% on 80% of the variants. Moreover, the combination of the explanatory power of DEOGEN2 in terms of evolutionary and contextual features, and of SNPMuSiC in terms of structure and stability, improves the contextualization of the impact that a mutation has at the protein level. For example, highly conserved residues located in the protein core, whose variants are predicted as deleterious by both DEOGEN2 and SNPMuSiC, are highly likely to be destabilizing, thus inducing (partial) unfolding of the protein. A full description of these predictors, their performance, large-scale applications and case studies related to the Niemann–Pick disease is available from Supplementary Material.
The new version of the MutaFrame server also provides additional computational and visualization utilities that help the users in the interpretation of the prediction results:
Visualization of the experimental or modeled 3D structure of the wild-type target protein, if available, and of the localization of the variant residue.
Per-residue solvent accessibility and secondary structure as well as additional information on the 3D protein structures such as the resolution of the X-ray structure or of the template used for the homology modeling.
DEOGEN2 and SNPMuSiC prediction scores of specific variants introduced by the user.
Heatmap showing the DEOGEN2 and SNPMuSiC scores of all possible variants in a target protein, both along the sequence and in the 3D protein structure.
Influence of the different features (residue conservation, protein essentiality,….) in the DEOGEN2 prediction.
Mapping between gene, protein sequence and protein structure identifiers and corresponding sequence alignments, for the entire human proteome.
Note, moreover, that all the results available on the webserver can easily be downloaded for offline analyses. In summary, MutaFrame facilitates the analysis of human variants at the molecular, evolutionary and contextual levels, thus going beyond the simple binary deleterious/benign classification. This constitutes an important asset in the clinical and biopharmaceutical fields.
Funding
This work was supported by the European Regional Development Fund and Brussels-Capital Region-Innoviris within the framework of the Operational Programme 2014–2020 [ERDF-2020 project ICITY-RDI.BRU]. F.P. and M.R. are post-doctoral Researcher and Research Director, respectively, at the F.R.S.-FNRS Fund for Scientific Research.
Conflict of Interest: none declared.
Acknowledgements
The authors thank I. Tanyalcin for his help in the technical setup of the web server.


{kind=link}