





Abstract
Multi-omics data in molecular biology has accumulated rapidly over the years. Such data contains valuable information for research in medicine and drug discovery. Unfortunately, data-driven research in medicine and drug discovery is challenging for a majority of small research labs due to the large volume of data and the complexity of analysis pipeline.
We present BioVLAB-Cancer-Pharmacogenomics, a bioinformatics system that facilitates analysis of multi-omics data from breast cancer to analyze and investigate intratumor heterogeneity and pharmacogenomics on Amazon Web Services. Our system takes multi-omics data as input to perform tumor heterogeneity analysis in terms of TCGA data and deconvolve-and-match the tumor gene expression to cell line data in CCLE using DNA methylation profiles. We believe that our system can help small research labs perform analysis of tumor multi-omics without worrying about computational infrastructure and maintenance of databases and tools.
http://biohealth.snu.ac.kr/software/biovlab_cancer_pharmacogenomics.
Supplementary data are available at Bioinformatics online.
1 Introduction
As the sequencing technologies advance rapidly, many database resources have been constructed, collecting and organizing molecular-level data from tumors and cancer cell lines. The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network, 2013) is a representative data resource for the multi-omics study of cancer biology. Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019) is a major database resource for cancer drug response. Analyzing the databases will bring us a wonderful opportunity to study cancer pharmacogenomics at the molecular level. However, there are several hurdles. First, analysis of multi-omics data requires use of computational tools and utilization of large-size databases such as TCGA and CCLE. Second, there is a significant gap between tumor and cell lines. Multi-omics data in TCGA are measured by bulk-cell sequencing of tumor that consists of different cell types such as immune cells and normal cells. On the other hand, investigation of cancer drug response in CCLE is primarily performed using cancer cell lines. Thus, integration of TCGA and CCLE is technically challenging. Another challenge is the availability of ‘computing resources’ where required computational tools and cancer databases are deployed.
2 Multi-omics data on the cloud
To address these challenges, we developed a bioinformatics system, BioVLAB-Cancer-Pharmacogenomics, on Amazon Web Services (AWS) (Supplementary Note S1). The main reasons why we deploy this system on AWS are as follows. First, Amazon provides TCGA and CCLE as pre-installed data resources on their cloud system. Second, AWS is the most widely used IaaS (Infrastructure as Service) that has been used by many people around the world. Third, analysis of multi-omics data requires a number of computational tools that should be orchestrated as a pipeline or workflow. Users have difficulty identifying computational tools thus installing and pipelining multiple tools to perform biologically meaningful analysis (Oh et al., 2021). We have several successful deployments of bioinformatics systems on Amazon as BioVLAB (Supplementary Note S2) (Chae et al., 2015; 2016; Lee et al., 2012). The main motivation of BioVLAB is to provide SaaS (Software as Service) on the IaaS cloud so that the user can use the bioinformatics system most conveniently without worrying about hardware and software resource management in a private computing space.
Figure 1 depicts the overview of our system. Once the user submits tumor multi-omics data and AWS credentials to the system via a web interface of front layer, all configuration and computation are done automatically in an ‘invisible’ area of AWS. The middle layer configures private computing space for the user. The multi-omics data, including single nucleotide variations (SNVs), somatic copy number alterations (SCNAs), DNA methylation and gene expression, are uploaded as input to pre-configured pipeline of the back layer (Supplementary Fig. S1). Based on the result of the algorithms, two reports are generated as output: intratumor heterogeneity (ITH) and pharmacogenomics (PG). ITH report shows the most closely related TCGA sample in terms of multi-omics heterogeneity (Carter et al., 2012; Park et al., 2016; Roth et al., 2014), as well as the relevant demographic, administration protocol, prognostic and treatment information (Supplementary Note S3). PG report shows the predicted sensitivity of the given tumor to various anticancer drugs by matching the bulk tumor to cell lines. We exactly followed the epigenomic deconvolution procedures described in the original EDec manuscript (Onuchic et al., 2016), which utilized 391 informative CpG loci that distinguishes four reference cell types (cancer epithelial, normal epithelial, stromal and immune) to deconvolve TCGA-BRCA expression profiles into the combination of eight cellular subpopulations. Then, we reconstructed pure cancer profile for the bulk tumor using only the cancer epithelial ones. (Supplementary Note S4, Supplementary Fig. S2).
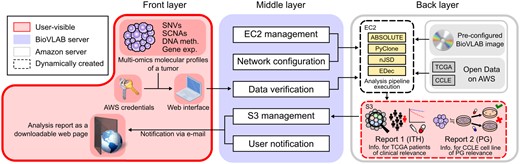
Overview of three layers in BioVLAB-Cancer-Pharmacogenomics. The front layer is the only layer that interacts with users. A user can submit cancer multi-omics data and AWS credentials of their own through the user interface in the front layer. The middle layer receives the input data from the front layer, verifies the validity of the data, and dynamically creates AWS EC2 instance from AMI image with pre-configured computing environment, and transfers data to the environment. The middle layer also dynamically creates S3 bucket and uploads the report once the analysis is done. The back layer consists of configured AWS EC2 instance and S3 bucket. Here is where the actual data analysis is performed. When the computation is done, the EC2 instance is automatically terminated to prevent overcharge. Boxes highlighted in red are only visible to the endpoint user. SNV, Single nucleotide variation; SCNA, Somatic copy number alteration; ITH, Intratumor heterogeneity; PG, Pharmacogenomics
Despite the complicated process, the user does not have to understand how the pipeline works because the whole analysis pipeline is encapsulated with straightforward user-level interfaces (Supplementary Fig. S3). The tasks for the user to analyze the data in our pipeline is kept to the minimum.
3 Case study using TCGA and CCLE
We demonstrate the usefulness of our approach by showing the experimental results that support the validity of each step of the system with TCGA-BRCA samples and CCLE-BRCA cell lines. Specifically, we investigate the utility of epigenomic deconvolution in the context of measuring the similarities between tumor samples and cancer cell lines. We also show that an array of cell lines in close relation to a given tumor sample helps suggesting an optimal drug treatment for the patient, as well as the utility of multi-omics ITH as a clinical biomarker.
To test whether the cancer-specific gene expression profile extracted from a bulk tumor represents the cancerous characteristics of a tumor better, we first examined how the similarities between the gene expression profiles of tumors and CCLE cancer cell lines change after the epigenomic deconvolution. The distribution of the pairwise cosine distances between standardized TCGA and CCLE gene expression profiles showed increased dispersion after the epigenomic deconvolution (Supplementary Fig. S4A). It implies that the deconvolution of bulk profiles disambiguates the mixed expression profiles and produces more precise signals of sample-specific cancer cells, which are closer to a specific subset of cell lines, as illustrated in (Supplementary Fig. S4B). Meanwhile, bulk tissue samples suffer from the transcriptomic noise originating from contaminating cells, resulting in more homogeneous distribution of pairwise distances with cancer cell lines. To support this argument further, the biological similarity between cancer expression profiles and top K nearest neighbors were examined in terms of PAM50 breast cancer subtypes. The top K nearest neighbors of deconvolved cancer expression profiles had more subtype-matching cell lines (Supplementary Fig. S4C). Similarly, cancer-specific expression profiles clarified the distinction of PAM50 breast cancer subtypes in the principal component space, both qualitatively (Supplementary Fig. S4D) and quantitatively (Supplementary Fig. S4E). This results collectively suggest that the epigenomic deconvolution purifies the cancer-specific expression profiles and it helps pinpointing specific cell lines that have similar molecular characteristics to the given cancer sample. To scrutinize it in more detail, we compared the distribution of pairwise cosine distances of PAM50 subtype-matching and subtype-mismatching TCGA-BRCA samples for each CCLE cell line and found that the differences between subtype-matching and subtype-mismatching distances were markedly increased after the deconvolution (Supplementary Fig. S4F).
We asked whether the similarity between purified tumor samples and CCLE cell lines can eventually be used to suggest improved response rates of patients for drug treatments. To this end, the clinical information for TCGA-BRCA harboring drug treatments and responses were analyzed retrospectively, using the publicly available drug responses (in log2 fold-change of cell viability) for CCLE cell lines from the primary screen of PRISM repurposing. Assuming the average drug responses of top 10 similar cell lines as an expected drug responses of a tumor sample, we can derive a set of suggested drugs that would elicit the best response for each specific patient. Specifically, a set of drugs giving rise to the average log fold-change of less than -2 were determined as suggested drugs. The rates of incomplete remissions showed moderate but meaningful differences between two groups of TCGA-BRCA patients, divided by whether or not they had been treated by suggested drugs (Supplementary Fig. S5). It supports that the experimentally characterized drug responses of similar cell lines can be effectively utilized to optimize the drug treatment for individual patient. As data accumulates from more cell lines in CCLE, this strategy can be more effective.
To illustrate the benefit of the simultaneous characterization of multi-omics ITH of a tumor, we compared the overall survival between the two TCGA-BRCA patient groups divided by the severity of various ITH measures. We used tITH measured by nJSD and genomic ITH (gITH) measured by PyClone as single-omics ITHs. The levels of multi-omics ITH (moITH) were determined by taking the geometric mean of the ranks of the two ITH measures. By examining the overall survival of 241 TCGA-BRCA patients who had complete clinical informations, we found that moITH had better predictive potential for the outcome than single-omics ITH measures (Supplementary Fig. S6). The result suggests that ITH levels from different omics layers are in part independent from each other, by which we can envision omics-specific mechanisms of ITH development. Thus, the integrative use of multi-omics ITH would complement the missing portion of ITH that could not be captured by single-omics analysis, resulting in a more thorough snapshot of ITH in a tumor sample.
4 Conclusion
BioVLAB-Cancer-Pharmacogenomics operates through a web-based interface to the cloud computing system for analyzing bulk cancer tissue multi-omics data. By providing the web interface, the usability of our system is significantly increased because the user can conveniently do the configuration of the cloud system via the web interface. This way, the user needs to use only the web browser for initiating the analysis of multi-omics data and email for receiving the analysis result. Our system is one of the first bioinformatics systems to utilize TCGA and CCLE on the AWS Open Data.
The utility of our system is demonstrated in three ways. First, to show that the deconvolved cancer profile of bulk tumor reflects well the pure cancerous aspect of it, we examined the cosine distances between the gene expression of TCGA samples and that of CCLE cell lines, before and after the deconvolution, and showed that the deconvolution helps identifying biologically relevant cell lines. Next, to show the effectiveness of anticancer drug suggestion in terms of PRISM cell viability tests on CCLE, we retrospectively examined the drug treatment done in TCGA-BRCA patients. As a result, the patients treated with drugs suggested by our system showed better complete remission (CR) rate. Lastly, we illustrated the benefit of ITH characterization in a multi-omics integrative way. A good use of multi-omics ITH as a predictive biomarker for patient survival is illustrated by the comparison with single-omics-based ITHs.
Our work provides a platform to explore multi-omics ITH and to match tumor and cell lines in TCGA and CCLE. Participation of many small research labs will be very useful to explore these important cancer biology questions.
Acknowledgement
The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga, and Broad Institute: https://portals.broadinstitute.org/ccle.
Funding
This research was supported by the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) [NRF-2014M3C9A3063541], the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea [HI15C3224], the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science & ICT [NRF-2019M3E5D4065965], and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science & ICT [NRF-2019M3E5D307337511].
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Sungjoon Park and Dohoon Lee authors should be regarded as Joint First Authors.


{kind=link}