




Abstract
Missense mutations in the leucine rich repeat kinase 2 (LRRK2) gene result in late-onset Parkinson’s disease. The incomplete penetrance of LRRK2 mutations in humans and LRRK2 murine models of Parkinson’s disease suggests that the disease may result from a complex interplay of genetic predispositions and persistent exogenous insults. Since neuroinflammation is commonly associated with the pathogenesis of Parkinson’s disease, we examine a potential role of mutant LRRK2 in regulation of the immune response and inflammatory signalling in vivo. Here, we show that mice overexpressing human pathogenic LRRK2 mutations, but not wild-type mice or mice overexpressing human wild-type LRRK2 exhibit long-term lipopolysaccharide-induced nigral neuronal loss. This neurodegeneration is accompanied by an exacerbated neuroinflammation in the brain. The increased immune response in the brain of mutant mice subsequently has an effect on neurons by inducing intraneuronal LRRK2 upregulation. However, the enhanced neuroinflammation is unlikely to be triggered by dysfunctional microglia or infiltrated T cells and/or monocytes, but by peripheral circulating inflammatory molecules. Analysis of cytokine kinetics and inflammatory pathways in the peripheral immune cells demonstrates that LRRK2 mutation alters type II interferon immune response, suggesting that this increased neuroinflammatory response may arise outside the central nervous system. Overall, this study suggests that peripheral immune signalling plays an unexpected—but important—role in the regulation of neurodegeneration in LRRK2-associated Parkinson’s disease, and provides new targets for interfering with the onset and progression of the disease.
Introduction
Pathogenic missense mutations in the leucine rich repeat kinase 2 (LRRK2) gene (formerly known as PARK8), result in an autosomal dominant late-onset Parkinson’s disease with clinical and pathological phenotypes similar to idiopathic Parkinson’s disease (Zimprich et al., 2004). In most cases, LRRK2-associated Parkinson’s disease is difficult to distinguish clinically from sporadic forms of the disease in terms of age of onset, disease progression and motor symptoms, including tremor, rigidity, postural instability and bradykinesia (Haugarvoll et al., 2008; Healy et al., 2008; Haugarvoll and Wszolek, 2009). In terms of pathology, both sporadic- and LRRK2-induced Parkinson’s disease are characterized by the preferential loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), the presence of intracellular α-synuclein-positive inclusions (Lewy bodies) in surviving SNpc dopaminergic neurons and decreased dopamine in the striatum (Wider et al., 2010).
LRRK2 is a large, complex protein containing six functional domains, two of which have known enzymatic activities; a ROC domain (GTPase activity) and a MAPKK domain (kinase activity). There are numerous single-nucleotide substitutions in each of these domains that have been shown to be pathogenic (West et al., 2005, 2007), including: G2019S, R1441G/C/H, Y1699C, I2020T, N1437H, G2385R, I2020T, I2012T and R1941H (Funayama et al., 2005, 2006; Khan et al., 2005; Lu et al., 2005; Haugarvoll and Wszolek, 2009; Satake et al., 2009; Bekris et al., 2010; Ross et al., 2011; Puschmann et al., 2012). The G2019S mutation located within the MAPKK domain is the most common cause of familial Parkinson’s disease, found in up to 41% of the Parkinson’s disease cases in North African Arab populations (Lesage et al., 2006; Kumari and Tan, 2009), and 18% of Ashkenazi Jewish Parkinson’s disease patients (Ozelius et al., 2006). The second most prevalent mutation, R1441G, located in the GTPase ROC domain, is present in about 46% of familial Parkinson’s disease that has an origin in the Basque region in Spain (Gaig et al., 2006; Gorostidi et al., 2009). In addition, LRRK2 mutations have been found in 5–10% of sporadic cases (Healy et al., 2008), suggesting that the mutations are linked to a common aetiology of the disease (Kumari and Tan, 2009).
In vitro studies demonstrated that Parkinson’s disease-linked mutations in the LRRK2 gene result in increased kinase activity, which is correlated with enhanced neuronal toxicity (West et al., 2005; Greggio et al., 2006; Luzon-Toro et al., 2007). In fact, toxic effects of mutant LRRK2 overexpression have been shown to be attenuated by pharmacological inhibition of LRRK2 kinase activity (Lee et al., 2010). Additionally, kinase-dead LRRK2 was shown to be non-toxic to neurons (Greggio et al., 2006; Smith et al., 2006; Skibinski et al., 2014). Based on these studies, it has been proposed that the pathological mechanism in LRRK2 Parkinson’s disease could be driven by a gain of function that results in increased kinase activity (Greggio et al., 2006). Initial in vitro studies using artificial substrates reported that only one mutation (G2019S) increases kinase activity, while other Parkinson’s disease-associated mutations (R1441G/C/H, I2020T, Y1699C) did not appear to have any significant effect on kinase activity (Jaleel et al., 2007; Nichols et al., 2010). However, recent in vivo studies have shown that LRRK2 mutations lying outside of the MAPKK domain (i.e. R1441G) can also lead to increased autophosphorylation (Sheng et al., 2012) as well as phosphorylation of a number of proteins, including those in the Rab family (West et al., 2005; Steger et al., 2016).
Although LRRK2 mouse models have shown impaired dopaminergic neurotransmission (Li et al., 2009; Tong et al., 2009), autophagic and mitochondrial abnormalities, and reduced neurite complexity (Ramonet et al., 2011), the majority of studies examining LRRK2 transgenic or knock-in mice do not report any apparent histopathological changes including the selective loss of dopaminergic neurons nor motor dysfunction (Tong et al., 2009; Li et al., 2010; Melrose et al., 2010; Herzig et al., 2012; Maekawa et al., 2012; Longo et al., 2014). The lack of manifestation of the most important Parkinson’s disease hallmarks in these murine LRRK2 models, along with the evidence from epidemiological studies of incomplete penetrance of LRRK2 mutations in Parkinson’s disease patients (Bonifati, 2007; Goldwurm et al., 2007; Latourelle et al., 2008; Xiromerisiou et al., 2012), suggest that other genetic factors or specific environmental insults might be required for mutant LRRK2 to trigger nigral cell loss (Lee and Cannon, 2015).
One of the most striking hallmarks of Parkinson’s disease, observed in both sporadic and familial LRRK2 patients, is a neuroinflammation characterized by increased cytokines level in the brain and CSF, extensive microglial activation, and infiltration of peripheral immune cells into the brain’s parenchyma (Rentzos et al., 2007; Brodacki et al., 2008; Brochard et al., 2009; Dufek et al., 2009; Hofmann et al., 2009; Deleidi and Gasser, 2013). Several in vitro studies demonstrated that wild-type LRRK2 expression is induced upon IFN-γ stimulation in peripheral immune cells and in BMDMs after exposure to microbial pathogens (Gardet et al., 2010; Hakimi et al., 2011). Additionally, wild-type LRRK2 has been suggested to activate NF-κB signalling in HEK293T cells (Gardet et al., 2010). Furthermore, LRRK2 knockout rats are partially protected from neurodegeneration induced by α-synuclein overexpression or intranigral lipopolysaccharide (LPS) exposure, suggesting that inhibition of LRRK2 with kinase inhibitors might have a neuroprotective effect in Parkinson’s disease (Daher et al., 2014). However, previous studies have not examined the role of pathogenic missense LRRK2 mutations in the peripheral and/or central immune response, glial activation and their role in subsequent neuronal death. Furthermore, while a growing amount of evidence suggests that many neurodegenerative diseases have an inflammatory component, the initiating factor(s) and whether these signals arise inside or outside of the CNS are not known. Therefore, in this study we sought to determine whether LRRK2 dysfunction induces a cross-talk between the periphery and the CNS immune systems to mediate both neuroinflammation and the degeneration of dopaminergic neurons. We triggered the innate immune response in mice overexpressing the two most common pathogenic LRRK2 mutations with systemic administration of an endotoxin, LPS, and analysed the neurodegenerative processes and development of immune response in both the CNS and periphery. These studies expand previous observations made in vitro or in LRRK2 knockout mouse models by demonstrating that acute endotoxaemia in LRRK2 mutant mice causes long-term neuronal loss and an exacerbated immune response in the brain compared to wild-type mice. However, the accelerated neuroinflammation in mutant mice is not triggered by dysfunctional microglia or invaded T cells and/or monocytes, but is mediated by circulating inflammatory molecules that are dysregulated by mutant LRRK2.
Materials and methods
Animals and LPS administration
In this study, 2-, 3-, 12- and 24-month-old FVB/NJ, C57BL/6J, FVB/N-Tg(LRRK2*R1441G)135Cjli/J, FVB/N-Tg(LRRK2)1Cjli/J (WT-OX), B6;C3-Tg(PDGFB-LRRK2*G2019S)340Djmo/J, C57BL/6J-Tg(LRRK2*R1441G)3IMjff/J and C57BL/6J-Tg(LRRK2*G2019S)2AMjff/J (all Jackson Laboratory) mice were used. In all experiments, age- and background strain-matched littermate mice were used as wild-type controls for transgenic strains. All animals were housed within the vivarium at St. Jude Children’s Research Hospital or Thomas Jefferson University and maintained on a 12:12-h light/dark cycle with ad libitum food and water. All of the experimental procedures in the animals were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and all protocols, were approved by the St Jude Children’s Research Hospital (Protocol 270) or Thomas Jefferson University (Protocol 1892) IACUCs. Experiments were carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for animal experiments.
Two- and 3-month-old mice were injected intraperitoneally with 0.9% sterile NaCl or LPS (5 mg/kg; Escherichia coli serotype O111:B4; 500 000 endotoxin units/mg) (Sigma-Aldrich). Warming of animals after injections was used to decrease hypothermic effects of the LPS (Romanovsky et al., 1997; Paul et al., 1999). All experimental procedures were performed under pathogen-free conditions.
Microglia isolation
Mouse adult microglia were isolated as described previously (Frank et al., 2006; Moussaud and Draheim, 2010; Lee and Tansey, 2013). The purity and the phenotype of microglia were verified by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot. For details see the online Supplementary material.
Single brain cell suspension preparation and FACS
Mouse adult brain tissue was dissociated for fluorescence-activated cell sorting (FACS) following the modified protocol of Brewer (Brewer and Torricelli, 2007). Samples were sorted for Thy+/CD3−, O4+ and GLAST+ (Schwarz et al., 2013; Sharma et al., 2015) using a FACSAria cell sorter (BD Biosciences). The purity of sorted cells was verified by western blot. For more details, see the online Supplementary material.
Peripheral blood mononuclear cell separation
Blood was collected from the heart of deeply anaesthetized mice into 10% EDTA-treated vacuum tubes, mixed 1:1 with sterile PBS and separated in lymphocytes separation medium (Corning). Samples were centrifuged for 30 min at 400g at room temperature and then the leucocyte ‘white’ layer was carefully aspirated and placed into sterile tubes. Cells were washed twice with three volumes of sterile PBS, centrifuged for 10 min at 200g at room temperature, and then stored at −80°C until assayed. For tandem mass tagging (TMT)-based proteomics experiments, leucocytes for each genotype and treatment were pooled from three to six mice.
Flow cytometry and FACS
For flow cytometric analysis, blood was collected from the heart of deeply anaesthetized mice into 10% EDTA-treated vacuum tubes. After red blood cell lysis with ammonium chloride solution, peripheral blood mononuclear cells (PBMCs) were stained with antibodies against CD4 (PerCP-Cy5.5), CD8 (PE-Cy7), CD19 (APC-Cy7), CD25 (APC), CD49 (PE), Mac-1 (Alexa700), Gr-1 (PerCP) (all from BD Biosciences), B220 (eFluor605; eBiosciences), and DAPI (Molecular Probes) for 30 min at 4°C. Data were acquired using a FACSCalibur flow cytometer (BD Biosciences) and analysed with CellQuest Pro software. For FACS, blood was pooled from 15 mice into 10% EDTA-treated vacuum tubes, and then the red blood cell-depleted PBMCs were incubated with anti-CD4 (APC), anti-CD8 (PerCP-Cy5.5), anti-CD11b (Alexa 700), anti-CD14 (PE), anti-CD19 (APC-Cy7) (all antibodies from BD Biosciences) and DAPI (Molecular Probes). Sorting of defined subpopulations was performed using a FACSAria cell sorter (BD Biosciences). For details see the online Supplementary material.
Cytokine assay
Cytokine expression in serum and brain samples was analysed with the Milliplex Map Kit, MCYTOMAG-70K (Millipore) and Luminex 200™. Data were analysed using BioPlex Manager 4.1 software. For details see the online Supplementary material.
Western blot
The following antibodies were used in this study: rabbit anti-LRRK2 (1:1000; clone 41-2, Abcam by MJFF), rabbit anti-pS935 LRRK2 (1:1000; Abcam by MJFF), rabbit anti-CD11b (1:1000; Abcam), rabbit anti-NeuN (1:1000; Abcam), mouse anti-GFAP (1:1000; Sigma-Aldrich), rabbit anti-MAP2 (1:500; Cell Signaling), mouse anti-GFAP (1:500; Cell Signaling), rabbit anti-MBP (1:500; Cell Signaling), rabbit or mouse anti-β-actin (1:2000; Abcam). For detailed description of the western blot protocol see the online Supplementary material.
RT-qPCR
Microglia, leucocytes and brain samples were processed to obtain RNA in accordance with the protocol outlined in PureLink® RNA Mini Kit or ARCTURUS® PicoPure® RNA isolation Kit (Thermo Fisher Scientific). Reaction was performed using TaqMan Gene Expression Master Mix or TaqMan® Fast Advanced Master Mix in a 7300 Real Time PCR System or 7500 Fast Real Time PCR System (Applied Biosystems), respectively. All samples were run in triplicate. For detailed description see the online Supplementary material.
Quantitative analysis of proteome by 10-plex TMT-LC/LC-MS/MS
Proteomic analysis was performed as detailed in Tan et al. (2017). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD006290 and PXD006289. For a detailed description see the online Supplementary material.
Immunohistochemistry
Immunostaining was performed as detailed in Jang et al. (2009), using antibodies directed against TH (mouse monoclonal, Sigma-Aldrich), Iba-1 (rabbit polyclonal, Wako; or goat polyclonal, Abcam), CD3 (Santa Cruz Biotechnology), Tmem119 (rabbit monoclonal; clone 28-3; provided by Prof. Ben Barres), and CCR2 (goat polyclonal, Thermo Fisher Scientific).
Stereological analysis
Stereological analysis of SNpc dopaminergic neurons and microglia were performed as detailed in Baquet et al. (2009).
Statistical analysis
All data are represented as mean ± standard error of the mean (SEM). Statistical analysis was performed using the GraphPad Prism® version 4.03 software. Differences between groups were determined by one-way ANOVA. If overall statistical significance was found, Tukey’s post hoc comparisons were performed.
Results
Characterization of LRRK2 mice
Since LRRK2 mutations result in late-onset Parkinson’s disease in humans (Zimprich et al., 2004), we first asked whether 2-year-old transgenic mice overexpressing Parkinson’s disease-linked LRRK2 mutations develop neuronal loss in the SNpc. We analysed two commercially-available transgenic strains overexpressing mutant human R1441G LRRK2 [FVB/N-Tg(LRRK2*R1441G)135Cjli/J and C57BL/6J-Tg(LRRK2*R1441G)3IMjff/J]; and two transgenic strains overexpressing mutant human G2019S LRRK2 [B6;C3-Tg(PDGFB-LRRK2*G2019S)340Djmo/J and C57BL/6J-Tg(LRRK2*G2019S)2AMjff/J]. Both strains overexpressing the human R1441G mutation were found to have the LRRK2 transgene inserted into mChr1, while the G2019S (340Djmo/J) and G2019S (2AMjff/J) strains have human LRRK2 transgene insertion sites in mChr3 and mChr2, respectively (Supplementary Fig. 1A). All strains analysed overexpress human mutant LRRK2 in the peripheral tissues and brain, including substantia nigra, striatum and cortex (Supplementary Fig. 1B). However, regardless of the strain background or the promoter used to drive mutant LRRK2, none of these four transgenic strains exhibited a loss of TH neurons in the SNpc or evidence of microgliosis at 2 years of age (Supplementary Fig. 1C and D). Two transgenic strains, R1441G (135Cjli/J) and G2019S (340Djmo/J), were used for the subsequent experiments (Supplementary Fig. 1E) since these have been previously characterized (Li et al., 2009; Ramonet et al., 2011; Mandemakers et al., 2012; Bichler et al., 2013; Sanchez et al., 2014; Mikhail et al., 2015).
Systemic inflammation induces dopaminergic-neuron loss in the substantia nigra pars compacta of mutant LRRK2 mice
Since transgenic mice overexpressing pathogenic LRRK2 mutations failed to develop an overt neurodegenerative phenotype—up to 2 years of age—we hypothesized that mutant LRRK2, acting as a stress-responsive kinase (Gardet et al., 2010; Hakimi et al., 2011; Lewis and Manzoni, 2012; Moehle et al., 2012), would be capable of facilitating neuronal loss only under specific conditions. Thus, we addressed a potential role for mutant LRRK2 in inflammation-induced neurodegeneration by evaluating the responses of mutant LRRK2 mice and their age-and strain-matched littermate wild-type controls to the administration of LPS (Supplementary Fig. 2A and B). To determine if LPS induces cell loss, we performed a stereological analysis of SNpc dopaminergic neurons (Baquet et al., 2009) from NaCl- and LPS-treated R1441G and strain-matched wild-type littermate controls 24 h, 7 days, 2 and 7 months post-LPS. We found that 24 h after LPS there was a small but significant loss of TH-positive neurons in the SNpc of R1441G mice while no change was observed in wild-type mice. At 7 days post-LPS, the loss of SNpc TH-positive neurons progressed, after which this loss stabilized (Fig. 1A and C). Nissl staining confirmed the loss of dopaminergic neurons in the SNpc of R1441G mice 7 months post-LPS (data not shown).
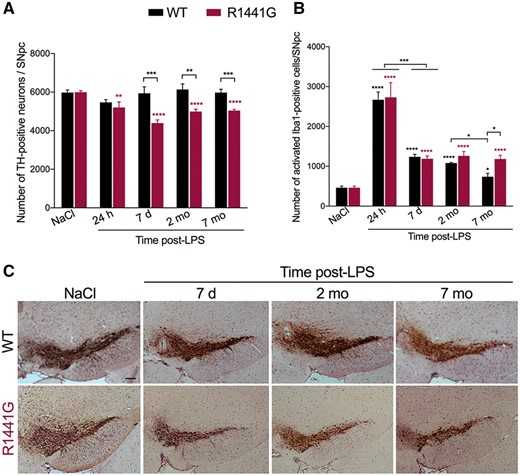
Systemic inflammation causes neuronal loss in the SNpc of mutant R1441G mice but not wild-type. (A) Loss of TH-positive dopaminergic neurons in the SNpc of R1441G mice up to 7 months (mo) after systemic LPS injection. Data are mean ± SEM, n = 16 for NaCl group, n = 3–5 for each post-LPS group. **P < 0.005, ***P < 0.001, ****P < 0.0001. (B) Number of activated Iba1-positive microglial cells in the SNpc of R1441G mice and background matched non-transgenic littermates (WT) following systemic LPS injection. Data are mean ± SEM, n = 15 for NaCl group, n = 3–5 for each post-LPS group. *P < 0.05, ***P < 0.001, ****P < 0.0001. (C) Representative images of TH-positive dopaminergic neurons in the SNpc of wild-type and R1441G mice 7 days (d), 2 and 7 months after LPS injection. Sections are matched at the same level of the substantia nigra (Bregma −3.08–3.16 mm) (Paxinos and Franklin, 2001). Scale bar = 100 μm.
To determine whether inflammation-induced neuronal loss is mutation specific, we exposed transgenic mice overexpressing mutant G2019S LRRK2 to LPS. Twenty-four hours after LPS, we found a significant loss of SNpc TH-positive dopaminergic neurons in the G2019S mice. The loss increased by 7 days after which the lesion became stable (Supplementary Fig. 2C and D). The loss of SNpc dopaminergic neurons was selective, as we observed no loss of glutamatergic pyramidal cells in the CA1 region of the hippocampus (NaCl: 39410 ± 618.5; LPS 7 months: 41867 ± 2063, n = 3; P = 0.31). Next, since both forms of mutant LRRK2 increased sensitivity of SNpc dopaminergic neurons to LPS, we examined if this effect was due to overexpression of LRRK2 itself or was specific to the mutant forms of the protein. When LPS was administered to mice overexpressing human wild-type LRRK2 (WT-OX) (Supplementary Fig. 2E and F), we observed no differences in the LPS-induced inflammatory response between wild-type and WT-OX (Supplementary Fig. 2G) or SNpc dopaminergic neuron loss (Supplementary Fig. 2H), suggesting that the neuron loss induced by LPS was dependent on mutant LRRK2.
Dopaminergic neuron loss in LRRK2 mice is not associated with the recruitment of peripheral immune cells into the brain parenchyma
Since recent studies have implicated the role of infiltrated peripheral immune cells, particularly T cells, in neuronal cell death (McGeer et al., 1988; Brochard et al., 2009; Cebrian et al., 2014), we next examined whether the loss of dopaminergic neurons seen in LPS-treated R1441G mice is mediated through invaded T cells. Here, coronal sections spanning the rostral-caudal extent of the brain were immunostained for a T cell marker, CD3. However, we found no evidence for T cell infiltration either in the substantia nigra or any other brain region of R1441G mice 24 h to 7 months post-LPS (Supplementary Fig. 3A). Immunohistochemical data were further confirmed by flow cytometric analysis of a single-cell brain suspension showing no CD3-positive cells in the LPS-treated R1441G brains (Supplementary Fig. 3B). Furthermore, immunostaining against a chemokine receptor, CCR2, which is known to be expressed on peripheral monocytes and macrophages but not in brain microglia (Mildner et al., 2007; Mizutani et al., 2012; Chiu et al., 2013), showed the lack of monocytes trafficking from the periphery (Fig. 2A). However, there was a significant increase in the number of activated Iba1-positive microglial cells in the SNpc following LPS injection with the number of Iba1 cells being higher in R1441G mice compared to wild-type mice 7 months post-LPS (Fig. 1B). Immunostaining with monoclonal antibodies that recognizes the microglia-specific transmembrane protein 119 (Tmem119) (Butovsky et al., 2014; Bennett et al., 2016) revealed that Tmem119 is co-localized with Iba1 following LPS treatment (Fig. 2B), suggesting that LPS-induced increase in the number of Iba1-positive cells resulted from a shift of microglial cells from a resting to an activated state rather than inducing infiltration of peripheral macrophages into the CNS.
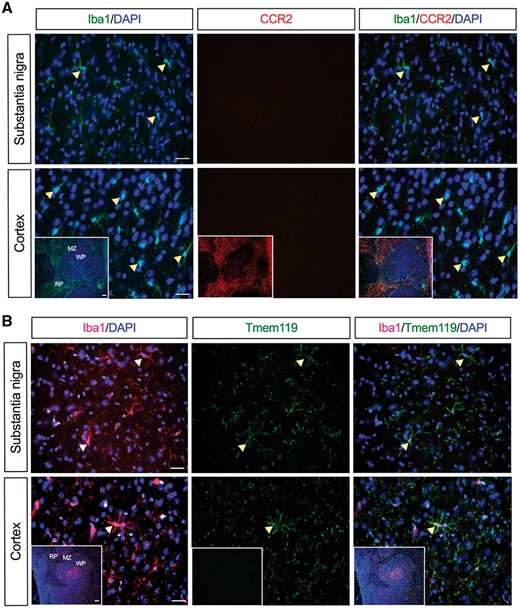
Lack of monocytes trafficking from the periphery into the brain of LPS-treated R1441G mice. (A) No CCR2 expression was seen in the substantia nigra and cortex of R1441G mice 24 h post-LPS. (B) Microglia-specific marker, Tmem119, is co-expressed in Iba1-positive microglia in the substantia nigra and cortex of R1441G mice 24 h post-LPS. Insets show spleen sections that were used as a positive and negative control for CCR2 and Tmem119 staining, respectively. Scale bar = 50 μm. Scale bar in insets = 100 μm. MZ = marginal zone; RP = red pulp; WP = white pulp.
Brain inflammatory profile is altered by mutant LRRK2
We investigated whether mutant LRRK2 alters the neuroinflammatory profile of the brain further. Thus, we assessed time-dependent changes in pro-apoptotic (TNF-α, IFN-γ, IL-1α), pro-inflammatory (IL-6) and anti-inflammatory cytokines (IL-10) as well as chemokines (CXCL-1, CXCL-10, CCL-2 (MCP-1), CCL-3 (MIP-1α) in the substantia nigra and striatum of normal and LPS-treated R1441G mice and their wild-type strain-matched littermate controls. At baseline, no significant differences in cytokine levels were observed between R1441G mice and their strain-matched littermate wild-type controls, except for small increases in CCL-2 and IFN-γ in the substantia nigra and a slight decrease in the levels of IL-10 and IL-1α in the striatum (Fig. 3A and B). However, following administration of LPS, significant differences were detected in the induction of IL-1α, IFN-γ, CXCL-1 and CXCL-10 in the substantia nigra and striatum of R1441G mice compared to wild-type controls, suggesting a generalized effect of mutant LRRK2 on the brain neuroinflammatory profile (Fig. 3C and D). Importantly, we found that IL-1α and IFN-γ, which have consistently been associated with neurodegeneration in vivo (Allan and Rothwell, 2001; Nesic et al., 2001; Allan et al., 2005; Mizuno et al., 2008; Pott Godoy et al., 2008; Chakrabarty et al., 2011; Mangano et al., 2012), remain elevated in the substantia nigra of R1441G mice even 2 months post-LPS compared to wild-type controls (Fig. 3C). To get a more detailed characterization of the altered neuroinflammatory reaction in the brain of LRRK2 mice, we performed mass spectrometry analysis (TMT) of brain tissue obtained from NaCl- and LPS-challenged wild-type (WT+LPS) and R1441G mutant mice (R1441G+LPS) (Fig. 4A). Mass spectrometry identified 10 743 proteins (<1% false discovery rate, FDR). For LPS-treated samples, differential expression analysis between R1441G+LPS and WT+LPS found 24 upregulated and 11 downregulated proteins that satisfied the power-based log2(FC) ≥ 0.37 and P ≤ 0.01 cut-off (Fig. 4B); and 137 upregulated and 73 downregulated proteins that satisfied the log2(FC) ≥ 0.22 and P ≤ 0.01 cut-off (Supplementary Table 1). None of the genes encoding the most enriched proteins in the R1441G group are located in the region of the transgene insertion on mChr1, suggesting that the transgene insertion itself is not a direct cause of the proteomic differences. Gene set enrichment analysis revealed several pathways that are preferentially enriched in the R1441G+LPS group compared to WT+LPS, including INF-γ response, IL6-JAK-STAT3 signalling, inflammatory response and TNF-α signalling (Fig. 4C and D). We next validated the expression of selected genes that encode proteins enriched in R1441G+LPS group in an independent experiment with a higher number of replicates and found that differences in mRNA expression for most genes in the R1441G+LPS group versus the WT+LPS group were similar to the one we observed for proteins in the proteomics experiment (Fig. 4E).
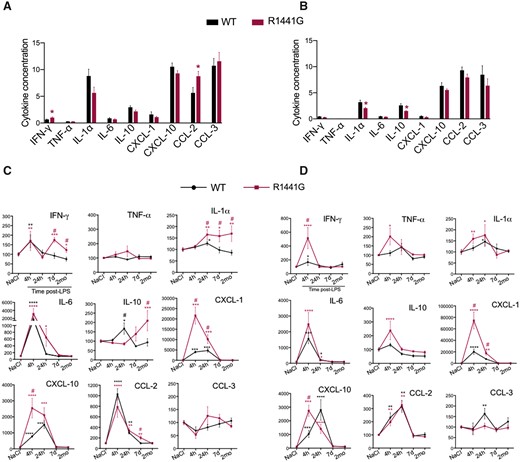
Mutant LRRK2 accelerates LPS-induced cytokine upregulation in the brain. (A and B) Cytokine profile of the substantia nigra (A) and striatum (B) of NaCl-injected R1441G mice and wild-type littermate controls. Data are expressed as pg/mg total protein. Data are mean ± SEM, n = 22–23. *P < 0.05 versus wild-type littermate controls. (C and D) Cytokines profile of the substantia nigra (C) and striatum (D) of R1441G mice and wild-type littermate controls at different time-points after LPS injection. Data are mean ± SEM, n = 15–16 for 24 h post-LPS, n = 5–8 for 4 h, 7 days, 2 months post-LPS. *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001 versus NaCl group; #P < 0.05 versus LPS-treated wild-type. For each genotype, data in LPS-treated groups are expressed as the percentage of NaCl control.
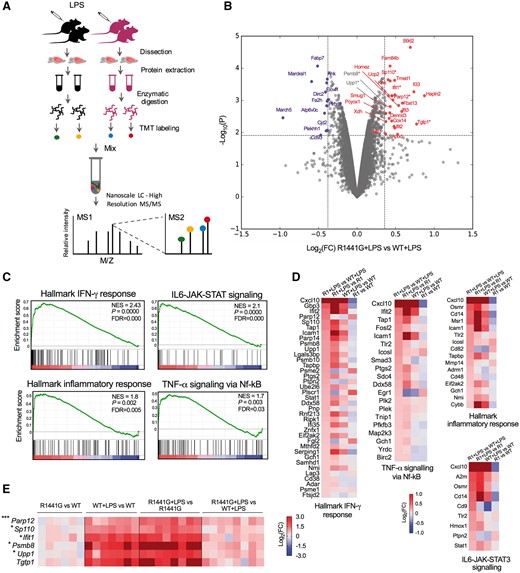
Brain inflammatory profile is altered by mutant LRRK2. (A) Experimental scheme for proteomic analysis of the brain samples. (B) Volcano plots of proteomic data from the brain of LPS-treated R1441G and wild-type littermate control mice. Log2fold-change(FC) is plotted across the x-axis and P-value is plotted on the y-axis. The red and blue nodes represent proteins upregulated and downregulated, respectively, in the R1441G+LPS group that satisfy log2(FC) ≥ 0.37 and P ≤ 0.01 cut-off. All differentially expressed proteins in the brain of LPS-treated wild-type and R1441G mice determined by quantitative proteomics analysis are listed in Supplementary Table 1. (C) GSEA plots and (D) heat maps showing upregulation of proteins involved in inflammatory response pathways in the brain of R1441G mice by comparison with littermate wild-type mice following in vivo LPS administration. Each bar at the bottom of the panel represents a member protein of the respective pathway. For each gene set enrichment assay, the nominal P-value, normalized enrichment score (NES) and FDR are shown. (E) Heat map showing mRNA expression of the selected genes in the brain of NaCl- and LPS-treated R1441G and wild-type littermate mice validated in an independent experiment. Target gene expression level was normalized to β-actin. Data are mean ± SEM, n = 6 for NaCl group; n = 7–8 for LPS group. *P < 0.05, ***P < 0.001, R1441G+LPS versus WT+LPS. Proteins encoded by validated genes and satisfy log2(FC) ≥ 0.37 cut-off are labelled with red asterisks in B. Other proteins encoded by validated genes are labelled with grey asterisks in B.
Exacerbated neuroinflammatory signalling in the brain of LRRK2 mice is not directly mediated through microglia
Given that microglial cells are the primary cells in the CNS capable of responding to the inflammatory stimulus, we next hypothesized that increased neuroinflammatory signalling and subsequent neuron damage in the SNpc of R1441G mice could be mediated through activated mutant microglial LRRK2. To test this hypothesis, we used well-described methods to isolate microglial cells from the brain (Cardona et al., 2006; Frank et al., 2006; Moussaud and Draheim, 2010), and then analysed LRRK2 expression and/or activation following the induction of systemic inflammation. RT-qPCR of isolated microglia confirmed the high purity of isolated cells (Fig. 5A). Furthermore, acutely isolated adult microglia from different brain regions expressed significantly elevated levels of pro-inflammatory markers upon in vivo activation with LPS compared to microglia from NaCl-treated mice (Fig. 5B), suggesting that isolated microglia can be used to reflect the LPS-induced inflammation-associated changes seen in vivo. However, subsequent western blot analysis of isolated microglia showed no LRRK2 expression or LPS-induced LRRK2 upregulation (Fig. 5C). These immunoblot data were confirmed by RT-qPCR showing almost no detectable Lrrk2 mRNA expression in microglia isolated from the midbrain of R1441G mice compared to the substantia nigra tissue (Fig. 5D). We also found no pS935 expression in isolated microglia (data not shown). Unexpectedly, these data for the first time suggest that ‘mutant’ microglia do not directly alter neuroinflammation in the brain of mice overexpressing pathogenic LRRK2. However, although not seen in the brain microglia, a significant and prolonged mutant LRRK2 upregulation (normal and phosphorylated) was found in the substantia nigra (Fig. 5E–G) and the whole brain tissue (Fig. 5H) of LPS-treated R1441G mice compared to LPS-treated strain-matched wild-type littermate controls. To determine what cells were responsible for the observed mutant LRRK2 upregulation, we used a FACS protocol (Brewer and Torricelli, 2007; Guez-Barber et al., 2012; Schwarz et al., 2013; Sharma et al., 2015) to purify neurons, astrocytes and oligodendrocytes from the adult mouse brain after in vivo stimulation with LPS (Supplementary Fig. 4A and B). Sorted cells were consistently >95% viable based on DAPI staining (Supplementary Fig. 4C). We confirmed the identity and purity of each sorted population (Thy1+/CD3− neurons, GLAST+ astrocytes, O4+ oligodendrocytes) using known cell type-specific proteins (Supplementary Fig. 4D). Further examination of sorted cell populations revealed that LRRK2 was highly expressed in the sorted Thy1+/CD3− neurons (Fig. 5I), and upregulated in neurons following LPS administration, but not in astrocytes or oligodendrocytes (Fig. 5J). Taken together, these data clearly suggest that ongoing inflammation has a long-term effect on intraneuronal mutant LRRK2 expression in vivo.
![Exacerbated neuroinflammation in the brain of LRRK2 mice is not mediated through microglia. (A) Normalized expression of Itgam, Gfap and Rbfox3 (NeuN) mRNA in isolated adult microglia from four different independent experiments was used to confirm the purity of isolated microglia. (B) Induction of inflammatory genes expression in acutely isolated adult microglia following in vivo LPS administration [n = 2; midbrain (Mb), striatum (Str) and cortex (Ctx) were pooled from three mice]. Inflammatory genes expression was normalized to the β-actin mRNA. Data are mean ± SEM. *P < 0.05 versus NaCl group. (C) Immunoblot of protein isolated from microglia obtained from NaCl- and LPS-treated R1441G mice. CD11b, GFAP and NeuN expression was used to confirm the purity of isolated microglia. (D) Normalized expression of mouse and human Lrrk2 mRNA in the substantia nigra and isolated midbrain microglia (Mg) from the brain of R1441G mice. Data are mean ± SEM; n = 3–6 (midbrain microglial cells were pooled from three R1441G mice, each pooled sample: n = 1). Lrrk2 mRNA expression levels were normalized to β-actin. (E–G) Total and pS935 LRRK2 upregulation in the substantia nigra following single LPS administration. Data are mean ± SEM; n = 19–20 for NaCl group, n = 5 for 4 h post-LPS, n = 9–11 for 24 h post-LPS, n = 6–10 for 7 days post-LPS, n = 4–5 for 2 months post-LPS; *P < 0.05, **P < 0.001, ***P < 0.0001. Total and pS935 LRRK2 protein levels were normalized to the level of β-actin. (H) Total LRRK2 upregulation in the whole brain tissue of R1441G mice 24 h following single LPS administration. (I) LRRK2 expression in sorted neurons (Thy1+/CD3−), astrocytes (GLAST+) and oligodendrocytes (O4+). Top two panels are different exposures of the same western blot. Relative band intensities of LRRK2 in each cell population were normalized to β-actin. Lrrk2−/− cortex sample was included as a negative control. (J) LRRK2 expression in sorted R1441G Thy1+/CD3− neurons, GLAST+ astrocytes and O4+ oligodendrocytes following in vivo LPS treatment. Relative band intensities of LRRK2 are normalized to β-actin. For each cell population fold change represents LRRK2 expression in LPS-treated group compared to NaCl control. (K) Lrrk2−/− brain samples were used to demonstrate anti-LRRK2 antibody specificity.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/141/6/10.1093_brain_awy077/6/m_awy077f5.jpeg?Expires=1716324581&Signature=0vS6s3mRrGkB7XZe-b4z2iUREmavYsQhBMS1Gls~NwQ13sWhG102HdF4nbH83UiMp~XAf6ES7bKfn~LdrUafSxNc1Bl-ibIIwq~xz4NYraiSJ1wYxF36LzqdLGEaCDQjKtN~F6c7EC4WHMYm6IGRI8f2-wjjvqdwtIML5qTrC79HkNrmNaod4ZWtRQ9260S9PytB4P2stvSmM7DmtOplcWNVMmxcfeV4OFfhd-A49U1~FX0hXSAW1xp8AM~Bsl9OIfdB8eqtK6V9Ptzkme4wk~YhHu74d8U5bzse7coBAklSxixZCrFeSxZRt4cSjnvHO3Y-L4IOAaFllSIFB1pOQw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Exacerbated neuroinflammation in the brain of LRRK2 mice is not mediated through microglia. (A) Normalized expression of Itgam, Gfap and Rbfox3 (NeuN) mRNA in isolated adult microglia from four different independent experiments was used to confirm the purity of isolated microglia. (B) Induction of inflammatory genes expression in acutely isolated adult microglia following in vivo LPS administration [n = 2; midbrain (Mb), striatum (Str) and cortex (Ctx) were pooled from three mice]. Inflammatory genes expression was normalized to the β-actin mRNA. Data are mean ± SEM. *P < 0.05 versus NaCl group. (C) Immunoblot of protein isolated from microglia obtained from NaCl- and LPS-treated R1441G mice. CD11b, GFAP and NeuN expression was used to confirm the purity of isolated microglia. (D) Normalized expression of mouse and human Lrrk2 mRNA in the substantia nigra and isolated midbrain microglia (Mg) from the brain of R1441G mice. Data are mean ± SEM; n = 3–6 (midbrain microglial cells were pooled from three R1441G mice, each pooled sample: n = 1). Lrrk2 mRNA expression levels were normalized to β-actin. (E–G) Total and pS935 LRRK2 upregulation in the substantia nigra following single LPS administration. Data are mean ± SEM; n = 19–20 for NaCl group, n = 5 for 4 h post-LPS, n = 9–11 for 24 h post-LPS, n = 6–10 for 7 days post-LPS, n = 4–5 for 2 months post-LPS; *P < 0.05, **P < 0.001, ***P < 0.0001. Total and pS935 LRRK2 protein levels were normalized to the level of β-actin. (H) Total LRRK2 upregulation in the whole brain tissue of R1441G mice 24 h following single LPS administration. (I) LRRK2 expression in sorted neurons (Thy1+/CD3−), astrocytes (GLAST+) and oligodendrocytes (O4+). Top two panels are different exposures of the same western blot. Relative band intensities of LRRK2 in each cell population were normalized to β-actin. Lrrk2−/− cortex sample was included as a negative control. (J) LRRK2 expression in sorted R1441G Thy1+/CD3− neurons, GLAST+ astrocytes and O4+ oligodendrocytes following in vivo LPS treatment. Relative band intensities of LRRK2 are normalized to β-actin. For each cell population fold change represents LRRK2 expression in LPS-treated group compared to NaCl control. (K) Lrrk2−/− brain samples were used to demonstrate anti-LRRK2 antibody specificity.
LPS-induced peripheral cytokines secretion and activation of immune pathways in leucocytes are exacerbated by mutant LRRK2
Based on the above experiments showing no LRRK2 expression and/or activation in brain microglia along with the evidence for lack of myeloid and T cell infiltration, we postulated that increased neuroinflammation in the brain of mutant mice could be mediated through circulating inflammatory mediators, and that the neurotoxic signals in mice carrying LRRK2 mutations might be generated outside the CNS. To test this hypothesis, we measured the protein level of peripherally-produced cytokines in the serum of wild-type and R1441G mice in response to LPS (Fig. 6A). First, we found that NaCl-injected wild-type or R1441G mice showed no genotype-related differences in cytokine concentration (Fig. 6B). However, following LPS administration, the expression wave of serum cytokines differed significantly starting 4 h after LPS; with increased levels of IFN-γ, IL-6, IL-10, CCL-5, G-CSF and M-CSF measured in R1441G mice compared to wild-type mice (Fig. 6C). To determine whether elevated cytokines affect LRRK2 expression in the peripheral immune cells, we isolated leucocytes from R1441G and wild-type littermate controls, and found that mutant LRRK2 upregulation was ∼10-fold greater in LPS-treated R1441G leucocytes than that in wild-type cells (Fig. 6D). We next carried out flow cytometric analysis (Supplementary Fig. 5A and B) to characterize peripheral mononuclear cell subpopulations in mutant LRRK2 mice and wild-type controls, and observed no significant difference in the number of lymphocyte subpopulations, macrophages and granulocytes between the two different genotypes after LPS exposure (Supplementary Fig. 5C). These data suggest that the upregulation in LRRK2 expression observed in LPS-treated mutant leucocytes are due to an increase in the expression of the protein on a per cell basis, and not to a different expansion of the number of mononuclear cells between the two genotypes. Subsequent FACS revealed that CD19+ B-lymphocytes are the main cells contributing to the increase in mutant LRRK2 expression (Fig. 6E). The observation that R1441G mice display a different peripheral cytokine profile in response to LPS next prompted us to determine the molecular pathways that might be affected by mutant LRRK2 in peripheral immune cells. To this end, we performed TMT-based proteomic analysis of the whole proteome of leucocytes isolated from non-treated and LPS-treated R1441G and wild-type mice (Fig. 6A). LRRK2 transgenic leucocytes and wild-type cells showed similar immune-response related protein expression in non-treated condition, however, differed significantly after the stimulation with LPS. Based on gene set enrichment analysis, we identified proteins in the IFN-γ response and inflammatory pathways (Fig. 6F and G) as being significantly enriched in leucocytes isolated from LPS-treated R1441G compared to LPS-treated wild-type mice. The highest overexpressed proteins in LPS-treated R1441G immune cells included antigen processing proteins (FCGR1, H2-Aa, TLR2), transcriptional factors (SP110, IRF8, IRF5, IRF9 and IRF4), mediators of apoptosis (GZMA, CASP1, CASP4, CASP8, PML), cytokines (IL18, CCL5), regulators of NF-κB (ICAM1, CD40, NFKB1, TRIM14) and MAPK signalling (FPR1) (Fig. 6G). Thus, mutant LRRK2 dysregulates LPS-induced activation of immune response related pathways in circulating mononuclear cells during the acute phase of inflammation.
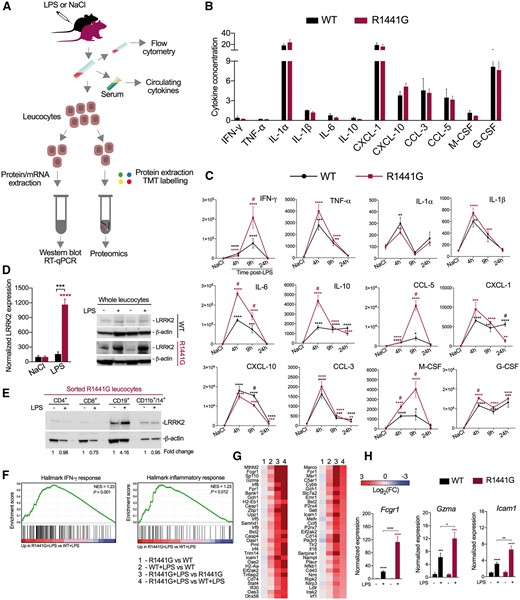
LPS-induced peripheral cytokines secretion and activation of immune pathways in leucocytes are exacerbated by mutant LRRK2. (A) Experimental scheme of peripheral immune response analysis. (B) Peripheral cytokines profile in the normal R1441G mice and wild-type littermate controls. Data are expressed as pg/mg total protein. Data are mean ± SEM, n = 5–8. (C) Inflammatory response in serum of LPS-treated R1441G mice compared to wild-type littermate controls. Data are mean ± SEM, n = 5–8 for NaCl and each post-LPS group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus NaCl group; #P < 0.05 versus LPS-treated wild-type. For each genotype data in LPS-treated groups are expressed as the percentage of NaCl control. (D) LRRK2 upregulation in leucocytes following in vivo LPS administration. Data are mean ± SEM, n = 4–5 for NaCl and 24 h post-LPS group. ***P < 0.001, ****P < 0.0001. (E) LRRK2 upregulation in sorted R1441G CD19+ B lymphocytes following in vivo LPS administration. For all groups, cells were pooled from 15 NaCl- or LPS-treated R1441G mice. (F) GSEA plots and (G) heat maps showing upregulation of IFN-γ response proteins and Inflammatory response proteins in isolated R1441G leucocytes by comparison with wild-type cells following in vivo LPS administration. Each bar at the bottom of panel in F represents a member protein of the respective pathway. For each gene set enrichment assay, the nominal P-value and normalized enrichment score (NES) are shown. (H) mRNA expression of the selected genes in the leucocytes isolated from NaCl- and LPS-treated R1441G and wild-type littermate mice validated in an independent RT-qPCR experiment. Target genes expression level was normalized to β-actin. Data are mean ± SEM, n = 4–5 for NaCl group; n = 3 for LPS group (for each n, cells were pooled from three mice). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Overall, the data in this study suggest that LPS-induced elevated neuroinflammation and neuronal loss in LRRK2 mutants are most likely initiated through circulating inflammatory mediators rather than a direct T cell or monocyte-mediated (both, microglia and macrophages) neurotoxicity.
Discussion
LRRK2 missense mutations are responsible for both familial and non-familial forms of late-onset Parkinson’s disease in humans (Zimprich et al., 2004); however, the precise mechanisms by which LRRK2 induces pathology are still unclear. Low penetrance of the mutations in humans (Bonifati, 2007; Goldwurm et al., 2007; Latourelle et al., 2008; Xiromerisiou et al., 2012), and the absence of an obvious neurological phenotype in LRRK2 mouse models (Tong et al., 2009; Li et al., 2010; Melrose et al., 2010; Herzig et al., 2012; Maekawa et al., 2012; Longo et al., 2014) suggest that Parkinson’s disease may result from a complex interplay of genetic predispositions and persistent exogenous insults. Causative factors include pathogens (bacterial or viral infections) (Puccini et al., 2015), protein aggregates (α-synuclein) (Deleidi and Gasser, 2013) or immune alterations associated with ageing or autoimmune disorders (Liu and Lenardo, 2012). In this study, we report new findings that expand our understanding by which LRRK2 mutations contribute to the development of Parkinson’s disease: (i) we demonstrate that LPS-induced inflammation is able to synergize with mutant LRRK2 (both R1441G and G2019S) to potentiate SNpc dopaminergic neuronal loss; (ii) we show that neurodegeneration is accompanied by an exacerbated neuroinflammation in the CNS, which has a subsequent effect on neurons by inducing intraneuronal LRRK2 upregulation; (iii) we provide evidence that enhanced neuroinflammation is not triggered by the brain’s resident microglia or infiltrated immune cells; and (iv) we show that mutant LRRK2 alters peripheral immune response and provides the first evidence of what inflammation related pathways could be modified by the pathogenic LRRK2 mutation.
One potential caveat to our findings is that these studies were carried out in mice that overexpressed mutant human LRRK2, rather than in mice carrying a knocked-in point mutation in the murine Lrrk2 gene (Herzig et al., 2011; Yue et al., 2015). We chose to use the human mutant LRRK2 BAC overexpression model for several reasons. First, knock-in G2019S mice develop neither neuronal loss/overt neuropathology nor abnormal locomotor response to dopaminergic agonists/antagonists (Herzig et al., 2011), suggesting that these knock-in LRRK2 mice do not phenocopy the human Parkinson’s disease pathology. Second, although the homology between the mouse and human LRRK2 gene is high [up to 94% in the ROC domain and 97% in the kinase domain (Langston et al., 2016)], it has been noted that there are significant differences in the gene’s regulatory regions (West et al., 2014). This lack of homology in the regulatory region could lead to differences in site(s) of protein expression as well as how the protein expresses in response to exogenous factors. For these reasons, we felt that use of the human BAC transgenic mice was an appropriate model for our studies. An additional caveat to our studies is that we cannot exclude the possibility that simply the overexpression of LRRK2 protein may, to some extent, alter the immune response; although have shown that LPS-induced inflammation in WT-OX mice does not result in neuronal loss, i.e. a parkinsonian phenotype. Additionally, recent studies found that WT-OX mice did not show statistically significant differences in peripheral monocyte subset ratios compared to R1441G mice (Bliederhaeuser et al., 2016), and the overexpression of wild-type LRRK2 had no effect on LPS-induced cytokine secretion by macrophages and on myeloid cell chemotaxis compared to non-transgenic cells (Moehle et al., 2015). Therefore, because of the lack of LPS-induced neurodegeneration in WT-OX mice, most experimental data on R1441G and/or G2019S were compared with background- and age-matched wild-type mice.
In this study, we do not observe LPS-induced SNpc dopaminergic neuron loss in wild-type mice; most likely due to the fact that the upregulation of pro-inflammatory mediators in the substantia nigra of wild-type mice is transient, returning to normal levels relatively quickly (within a week); i.e. the brain parenchyma is able to modulate the neuroinflammatory response. However, unlike what was observed in wild-type mice, LRRK2 mutant mice exhibited an exaggerated long-term microglial activation with extended secretion of inflammatory factors, including IFN-γ and IL-1α, that have been shown to mediate dopaminergic neuron degeneration (Pott Godoy et al., 2008; Chakrabarty et al., 2011; Mangano et al., 2012). The finding of enhanced SNpc dopaminergic neuron loss in the R1441G LRRK2 mutant mice subsequent to LPS exposure with continued cytokine/chemokine secretion suggests an ongoing process where inflammatory factors provoke a cycle of inflammation in the brain that eventually reaches a critical ‘threshold’, triggering a self-perpetuating cycle of inflammation and neurodegeneration. Critically, we showed for the first time that ongoing neuroinflammation leads to the upregulation of intraneuronal mutant LRRK2 in vivo, which may also subsequently contribute to neurotoxicity observed in mutant mice since intraneuronal LRRK2 level has been shown to be proportional to cell death (Skibinski et al., 2014).
Since microglia, because of its high expression of TLR4 (Zhou et al., 2006), are the primary cells capable of responding to the inflammatory stimulus in the brain, we hypothesized that neuroinflammation in the CNS of LRRK2 mice, especially in the substantia nigra, which is enriched with microglia (Lawson et al., 1990), could be directly exacerbated by the mutant microglial LRRK2. However, unlike recent findings showing wild-type LRRK2 upregulation after intranigral injection of LPS in isolectin B4+ cells with a morphology and size consistent with activated microglia (Moehle et al., 2012), we did not detect LRRK2 expression in microglia isolated from either whole brain or midbrain, both before and after LPS-induced inflammation. What could explain this difference? Bearing in mind that peripheral monocytes have detectable expression of LRRK2, and intracerebral, but not peripheral, LPS injection induces a massive extravasation of leucocytes into the brain parenchyma (Zhou et al., 2006; Ji et al., 2007), we postulate that that the LRRK2+/isolectin B4+ cells are not resident microglia, but most likely represent monocytes from the periphery that invade the CNS. This hypothesis is supported by recent studies from the West lab, who showed LRRK2 immunoreactivity in CD68/iNOS-positive cells recruited in response to α-synuclein and LPS-transduced SNpc (Daher et al., 2014).
The lack of LRRK2 expression in microglia has also been shown in numerous immunohistochemical and in situ hybridization studies on brain sections obtained from healthy controls and patients with Parkinson’s disease. These studies demonstrated that LRRK2 protein and/or mRNA was localized exclusively to neuronal perikarya and dendritic/axonal processes (Higashi et al., 2007; Hakimi et al., 2011; Sharma et al., 2011; Dzamko et al., 2012, 2017). Furthermore, microglia from adult wild-type mice and BAC transgenic LRRK2 mice do not show any LRRK2 immunoreactivity in vivo (Biskup et al., 2006; Higashi et al., 2007; Westerlund et al., 2008; Mandemakers et al., 2012). However, we recognize that the finding of a lack of LRRK2 expression in adult microglia is somewhat controversial. Indeed, initial in vitro studies examining this showed that a relatively high level of LRRK2 was expressed in mouse peripheral macrophages, monocytes (Gardet et al., 2010) and cultured bone marrow-derived macrophages (Hakimi et al., 2011). These findings led to the uniform assumption that LRRK2 is expressed in all cell types of myeloid origin, at any developmental stage. Others have shown that primary P2 rat microglia (Moehle et al., 2012) and the immortalized murine microglial cell line, BV-2 (Kim et al., 2012; Russo et al., 2015) also express LRRK2. However, this expression was seen in cultured and/or immortalized cells that have been shown to act substantially different from cognate CNS resident microglia (Schmid et al., 2009); including the significant finding that they do not express the molecular signature(s) seen in adult microglial cells (Butovsky et al., 2014).
Our results, in concert with previous studies that demonstrated no microglia-specific LRRK2 protein and/or mRNA expression in the brain sections from healthy controls and patients with Parkinson’s disease (Higashi et al., 2007; Hakimi et al., 2011; Sharma et al., 2011; Dzamko et al., 2012, 2017), support the hypothesis that ‘mutant’ microglia cannot directly alter neuroinflammation in the brain of mice overexpressing the parkinsonian mutation, and for the first time raise an important question of whether LRRK2 mutations impair peripheral immune cell function(s) with a deleterious impact on brain microglia and dopaminergic neurons as a secondary effect.
This leads us to propose that the exacerbated neuroinflammation measured in the CNS of mice carrying LRRK2 mutations occurs as a peripheral-to-centrally-mediated immune signal, and does not depend on the active participation of microglia or infiltrating T cells and monocytes. In fact, by monitoring circulating cytokine production and inflammatory signalling in peripheral leucocytes of LPS-challenged wild-type and mutant LRRK2 mice, we demonstrated that this Parkinson’s disease-causing mutation significantly dysregulates LPS-induced activation of peripheral cytokines and inflammatory proteins in the leucocytes, including IFN-inducible proteins (SP110, IRF8, IRF5, IRF9 and IRF4), antigen processing proteins (TLR2, FCGR1, H2-Aa) and crucial mediators of NF-κB and MAPK signalling (ICAM1, CD40, NFKB1, TRIM14, FPR1). Although the mechanisms connecting the peripheral immune response and neuroinflammation are not yet fully understood, it is known that blood-borne cytokines, including IL-1α, IL-1β, IL-6 and TNF-α can reach the brain and transduce the inflammatory response to relevant cell types through either ‘leaky’ regions in the blood–brain barrier located in the circumventricular organs (passive diffusion) or directly via saturable transport systems on endothelial cells (Banks et al., 1995; Pan et al., 2011). Once activated by blood-borne cytokines, both microglia and endothelial cells expressing cytokine receptors (Van Dam et al., 1996; Reyes et al., 1999; Verma et al., 2006) may subsequently amplify a cascade of local neurotoxic mediators even after resolution of the peripheral inflammation. However, microglia and endothelial cells in LRRK2 mutants seem to not be related to the accelerated neuroinflammatory response per se, but act more in a modulatory rather than primary role. Importantly, one should keep in mind that although the blood–brain barrier is almost impermeable to LPS (Nadeau and Rivest, 2002; Banks and Robinson, 2010), TLR4-positive cells were found along large brain vessels and within the choroid plexus, meninges, circumventricular organs, and the ventricular ependymal lining (Chakravarty and Herkenham, 2005). If endothelial cells possess the ability to express LRRK2, they might also alter the cascade of inflammatory signals initiated by peripherally administrated LPS, though unlikely since LRRK2 has been found so far only in human umbilical vein endothelial cells (Hongge et al., 2015), and little is known about its expression in brain microvascular endothelial cells.
Several clinical reports have reported on the association of alterations in the peripheral immune response associated with familial Parkinson’s disease to central inflammation and neurodegeneration. For example, the involvement of peripheral inflammation in LRRK2-associated Parkinson’s disease can be supported by studies demonstrating dysregulated interleukin and TGF-β signalling in immune cells from G2019S patients (Mutez et al., 2011), as well as the increase in peripheral pro-inflammatory cytokines in the asymptomatic G2019S carriers (Dzamko et al., 2016). Moreover, a genome-wide association study revealed a possible involvement of different single-nucleotide polymorphisms in the LRRK2 gene in Crohn’s disease, an inflammatory bowel disease (Franke et al., 2010), and leprosy, a chronic infectious disease caused by Mycobacterium leprae (Zhang et al., 2009; Fava et al., 2016). The widespread distribution of LRRK2 in peripheral tissues related to immune response and its genetic implication in inflammatory disorders such as Crohn’s disease and leprosy reinforce the notion that LRRK2 may act as a regulator of the immune system. Indeed, recent in vitro studies have demonstrated a robust IFN-γ-stimulated increase of endogenous wild-type LRRK2 expression across various subsets of cultured myeloid cells, lymphocytes, bone marrow-derived macrophages and transformed cell lines, including human THP-1 cells and murine RAW264.7 macrophage-like cells (Gardet et al., 2010; Hakimi et al., 2011; Gillardon et al., 2012; Kuss et al., 2014). However, our data clearly indicate that under in vivo stimulating conditions, mutant LRRK2 is solely upregulated in lymphocytes, an in particular B lymphocytes, and not in myeloid cell lineage (peripheral macrophages or CNS microglia), while wild-type LRRK2 seems to be unchanged in all types of cells. Given that in vivo interactions between immune cells are extremely dynamic and appear to be bidirectional, the physiological interactome of mutant LRRK2 in ‘authentic’ immune cells could be far more complex than those seen in vitro. Our observation that B lymphocytes display activated mutant LRRK2 over other immune cells under stimulated conditions along with the studies showing high LRRK2 expression in human B cells (Gardet et al., 2010; Hakimi et al., 2011; Thevenet et al., 2011) raises the intriguing question of whether B cell-derived LRRK2 could be involved in the regulation of immune response to pathogens through secreted antibodies or by cellular interactions and/or cytokine-dependent mechanisms. Therefore, future in-depth examinations of immune response and LRRK2 activation in B cells derived from idiopathic and LRRK2 Parkinson’s disease patients may help clarify the role of mutant LRRK2 in health and disease. In addition, further in vivo studies on transgenic mice lacking mutant LRRK2 expression in lymphocyte subsets with mutant LRRK2 expression in the nervous system are required to define the mechanism(s) connecting the peripheral immune response with neurodegeneration in the CNS.
Acknowledgements
We thank Drs Ben Barres and Mariko Bennett (Stanford University School of Medicine) for providing Tmem119 antibody; Dr Mark Cookson (Laboratory of Neurogenetics, National Institute of Aging, NIH) for providing Lrrk2−/− samples; Drs Mark Valentine and Virginia Valentine from the St. Jude Cytogenetic Core for performing FISH analysis; Drs Richard Ashmun and Stacie Woolard from St. Jude Flow Cytometry and Cell Sorting Shared Resource for assistance with Flow Cytometry and Cell Sorting; Dr Jamshid Temirov from St Jude Cell and Tissue Imaging Center for assistance with light microscopy and imaging.
Funding
Supported by Cancer Center Support Grant P30 CA021765, The American-Lebanese Syrian Associated Charity and Thomas Jefferson University.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- LPS
lipopolysaccharide
- SNpc
substantia nigra pars compacta
- WT-OX
mice overexpressing human wild-type LRRK2
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}