




Abstract
Axonal degeneration is an early and ongoing event that causes disability and disease progression in many neurodegenerative disorders of the peripheral and central nervous systems. Chemotherapy-induced peripheral neuropathy (CIPN) is a major cause of morbidity and the main cause of dose reductions and discontinuations in cancer treatment. Preclinical evidence indicates that activation of the Wallerian-like degeneration pathway driven by sterile alpha and TIR motif containing 1 (SARM1) is responsible for axonopathy in CIPN. SARM1 is the central driver of an evolutionarily conserved programme of axonal degeneration downstream of chemical, inflammatory, mechanical or metabolic insults to the axon. SARM1 contains an intrinsic NADase enzymatic activity essential for its pro-degenerative functions, making it a compelling therapeutic target to treat neurodegeneration characterized by axonopathies of the peripheral and central nervous systems. Small molecule SARM1 inhibitors have the potential to prevent axonal degeneration in peripheral and central axonopathies and to provide a transformational disease-modifying treatment for these disorders.
Using a biochemical assay for SARM1 NADase we identified a novel series of potent and selective irreversible isothiazole inhibitors of SARM1 enzymatic activity that protected rodent and human axons in vitro. In sciatic nerve axotomy, we observed that these irreversible SARM1 inhibitors decreased a rise in nerve cADPR and plasma neurofilament light chain released from injured sciatic nerves in vivo. In a mouse paclitaxel model of CIPN we determined that Sarm1 knockout mice prevented loss of axonal function, assessed by sensory nerve action potential amplitudes of the tail nerve, in a gene-dosage-dependent manner. In that CIPN model, the irreversible SARM1 inhibitors prevented loss of intraepidermal nerve fibres induced by paclitaxel and provided partial protection of axonal function assessed by sensory nerve action potential amplitude and mechanical allodynia.
Introduction
Axonal degeneration is an early pathophysiological manifestation of neurodegeneration in many degenerative diseases of the PNS and CNS characterized by dying-back pathology.1 This Wallerian-like axonal degeneration precedes death of the neuronal cell body, and involves a mechanism of axonal dismantling in response to pathological insults that trigger activation of sterile alpha and TIR motif containing 1 (SARM1) and its intrinsic NADase.2 This enzymatic activity is contained within the TIR domain of the molecule and is responsible, through molecular mechanisms still under investigation, for a local catastrophic bioenergetic crisis that initiates a cell autonomous process of axonal self-destruction.2 Current understanding of SARM1 function implicates the catalytic site of the NADase as both necessary and sufficient to initiate this process. Mutagenesis studies demonstrated that changing a single amino acid residue in the catalytic domain, E642A, is sufficient to protect axons and completely eliminate the SARM1-dependent injury response to a number of mechanical, chemical and metabolic insults.3 Consequently, as the central executioner of the Wallerian and Wallerian-like axonal degeneration programmes, activation of SARM1 and in particular its intrinsic NADase activity, has been implicated as a major therapeutic target for the treatment of axonopathies and neuropathies.4-6
Peripheral neuropathies represent the most common form of neurodegeneration and affect millions of people worldwide.7 Models of inherited neuropathies such as Charcot-Marie-Tooth, and several models of diabetic and chemotherapeutic induced neuropathies have shown benefit when inhibiting the Wallerian-like degeneration pathway.8–15 Chemotherapy-induced peripheral neuropathy (CIPN), a well-characterized and predictable form of peripheral neuropathy, results from exposure to various chemotherapeutic agents such as taxanes, vinca alkaloids and proteasome inhibitors such as bortezomib.10,16 This common pathology is the most frequent cause of discontinuations, dose reductions, dose skipping and dose limitation during cancer treatment.17,18 Since those events affect clinical outcome and survival, CIPN represents a major unmet medical need in oncology.
Recently, we demonstrated that a class of isoquinoline small molecule reversible inhibitors of the SARM1 NADase can protect axons in vitro from traumatic injuries and mitochondrial dysfunction.19 As a first step in developing SARM1 inhibitors to treat peripheral indications, we identified and developed a new class of orally bioavailable irreversible small molecule inhibitors of the SARM1 NADase and demonstrated their ability to protect axonal structure and function in vitro and in animal models of CIPN.
Materials and methods
Preparation of SAM-TIR lysate
NRK1-HEK293T cells3 were seeded onto 150-cm2 flasks at 10 × 106 cells per plate. The next day, the cells were transfected with 15 μg of human SAM-TIR expression plasmid using X-TremeGENETM 9 DNA Transfection Reagent. The human SAM-TIR expression plasmid consisted of Strep-TEV-human SARM1, amino acids 408–700, cloned into pSF-CMV-Amp using NcoI and XbaI sites. The cultures were supplemented with 1 mM nicotinamide riboside at the time of transfection to minimize toxicity from SAM-TIR overexpression.3 Forty-eight hours after transfection, cells were harvested, pelleted by centrifugation at 1000 rpm (Sorvall ST 16 R centrifuge, ThermoFisher) and washed once with cold PBS (0.01 M PBS; NaCl 0.138 M; KCl 0.0027 M; pH 7.4). The cells were resuspended in PBS with protease inhibitors (cOmpleteTM protease inhibitor cocktail) and cell lysates were prepared by sonication (Branson Sonifer 450, output = 3, 20 episodes of stroke). The lysates were centrifuged (12 000g for 10 min at 4°C) to remove cell debris and the supernatants (containing SARM1 SAM-TIR protein) were stored at −80°C for later use in the in vitro SARM1 SAM-TIR NADase assay. Protein concentration was determined by the bicinchoninic (BCA) method and used to normalize lysate concentrations.
SAM-TIR NADase assay
The enzymatic assay was performed as described previously.19 In brief, SAM-TIR lysate at a final concentration of 5 µg/ml was preincubated with the respective compound in Dulbecco’s PBS buffer at 1% dimethylsulphoxide (DMSO) final assay concentration in a final assay volume of 20 µl over 2 h at room temperature in a 384-well polypropylene plate. The reaction was initiated by addition of 5 µM final assay concentration of NAD+ as substrate. After a 2 h incubation at room temperature, the reaction was terminated with 40 µl of stop solution of 7.5% trichloroactetic acid in acetonitrile. The NAD+ and ADPR concentrations were analysed by online solid phase extraction coupled with tandem mass spectrometry (SPE-MS/MS) using a RapidFire 300 (Agilent Technologies) coupled to an API4000 triple quadrupole mass spectrometer (AB Sciex). A modified version of this assay was used to incubate SAM-TIR lysate to Strep-Tactin®XT-coated plates (IBA) to capture the Strep-tag moiety in the recombinant SARM1 construct. This modified assay was used to test the effect of compound removal on enzymatic activity. We incubated 30 µg/ml total protein from SAM-TIR lysates per well in Strep-Tactin®XT plates and rinsed to remove unbound lysate material. Plate-bound SAM-TIR protein was incubated with the compounds for 2 h and subsequently the plates were rinsed and incubated in assay solution without NAD+ for various times, as indicated in the text. At the end of the incubation time, plates were rinsed to remove any compounds that would have been released during the washout incubation period, and the enzymatic reaction was initiated by adding NAD+.
Screening of compound libraries
A collection of ∼200 000 diverse small molecules, representing a wide variety of chemotypes, was screened at a single concentration for the ability to inhibit NAD+ turnover by SARM1 in the SAM-TIR NADase assay. Hits from this initial high-throughput screen were validated in concentration response curves to provide well qualified tool SARM1 inhibitors and starting points for further optimization. The isothiazoles described in this study represent a chemical series chosen for optimization.
Selectivity and safety panel screening
The data in Table 1 were generated at BPS Bioscience (San Diego, CA). The full list of assays and associated methods can be found at the BPS Bioscience website (https://bpsbioscience.com/screening-and-profiling, accessed 3 October 2021).
Profiling of isothiazoles against cysteine mutants in the TIR domain
| Compound | Wild-type SAM-TIR IC50 (µM) | C649A SAM-TIR IC50 (µM) | C635A SAM-TIR IC50 (µM) |
|---|---|---|---|
| 4 | 0.37 | 0.57 | 12 |
| 8 | 1.2 | 1.2 | 17 |
| 9 | 0.16 | 0.46 | 2.4 |
| Compound | Wild-type SAM-TIR IC50 (µM) | C649A SAM-TIR IC50 (µM) | C635A SAM-TIR IC50 (µM) |
|---|---|---|---|
| 4 | 0.37 | 0.57 | 12 |
| 8 | 1.2 | 1.2 | 17 |
| 9 | 0.16 | 0.46 | 2.4 |
Profiling of isothiazoles against cysteine mutants in the TIR domain
| Compound | Wild-type SAM-TIR IC50 (µM) | C649A SAM-TIR IC50 (µM) | C635A SAM-TIR IC50 (µM) |
|---|---|---|---|
| 4 | 0.37 | 0.57 | 12 |
| 8 | 1.2 | 1.2 | 17 |
| 9 | 0.16 | 0.46 | 2.4 |
| Compound | Wild-type SAM-TIR IC50 (µM) | C649A SAM-TIR IC50 (µM) | C635A SAM-TIR IC50 (µM) |
|---|---|---|---|
| 4 | 0.37 | 0.57 | 12 |
| 8 | 1.2 | 1.2 | 17 |
| 9 | 0.16 | 0.46 | 2.4 |
Experimental animals
All animal experiments were carried out in accordance with the regulations of the German animal welfare act and the directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Protocols were approved by the local ethics committee of the Authority for Health and Consumer Protection of the city and state Hamburg (‘Behörde für Gesundheit und Verbraucherschutz’ BGV, Hamburg). Mice were housed on a 12-h light/dark cycle with ad libitum access to food and water.
For in vitro studies, tissue extraction and ex vivo cell culture experiments were carried out according to the regulations of the German animal welfare act and the directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and were approved by the city and state Hamburg (BGV, Hamburg).
Neuronal cultures
Mouse dorsal root ganglion (DRG) cultures were performed as described previously.19,20 Mouse DRGs were dissected from embryonic Day 13.5 C57BL/6J mouse embryos (50 ganglia per embryo) and incubated with 0.05% trypsin solution containing 0.02% EDTA (ThermoFisher) at 37°C for 15 min. Then, cell suspensions were triturated by gentle pipetting and washed three times with DRG growth medium (NeurobasalTM medium; ThermoFisher) containing 2% B27 (ThermoFisher), 100 ng/ml 2.5S NGF (Sigma), 1 μM uridine (Sigma), 1 μM 5-fluoro-2ʹ-deoxyuridine (Sigma), penicillin and streptomycin. Cells were suspended in DRG growth medium at a ratio of ∼100 μl medium/50 DRGs. The cell density of these suspensions was adjusted to 107 cells/ml. Cell suspensions (0.5 μl/well; 5000 cells/well) were placed in the centre of the well using 96-well tissue culture plates (Corning) coated with poly-d-lysine (0.1 mg/ml; Sigma) and laminin (3 μg/ml; ThermoFisher). Cells were allowed to adhere in a humidified tissue culture incubator (5% CO2) for 15 min and then DRG growth medium was gently added (200 μl/96 well). Axon degeneration assays19,20 were performed at days in vitro (DIV) 6–7. Unless otherwise indicated in the figures, cultures were pretreated with compounds for 2 h, rinsed twice with warm culture media, injured by manual axotomy using a micro-surgical blade and examined 16 h post-axotomy. Compounds were initially dissolved in DMSO to make a 10 mM stock solution, compound treatment was as indicated in the figures and final DMSO concentration in the cultures was 0.3%.
Human induced pluripotent stem cell-derived motor neurons
Human induced pluripotent stem cell (iPSC)-derived motor neurons (C1048) were obtained from Cellular Dynamics and grown as spotted cultures.19 Cells were thawed at in a 37°C water bath for 2 min 30 s and transferred to Complete Maintenance Medium (iCell Neural Base Medium 1, 2% iCell Neural Supplement A, 1% iCell Nervous System Supplement) + 5 µM (2S)-N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2-phenyl]glycine 1,1-dimethylethyl ester (DAPT), as described by the manufacturer, and cell density was adjusted to 10 000 cells/µl. Cell suspensions (1 μl/well; 10 000 cells/well) were placed in the centre of the well using 96-well tissue culture plates coated with poly-d-lysine (0.1 mg/ml; Sigma) and laminin (3 μg/ml; ThermoFisher). Cells were allowed to adhere in a humidified tissue culture incubator (37°C, 5% CO2) for 10 min and then Complete Maintenance Medium + 5 µM DAPT was gently added (200 μl/well). Cells were maintained in a humidified incubator at 37°C, 5% CO2 for 2 weeks with 75% of the media replaced three times/week during the first week with Complete Maintenance Medium + 5 µM DAPT and 50% of the media replaced three times/week with Complete Maintenance Medium during the second week.
Immunocytochemistry
DRG cultures were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min followed by gentle PBS rinse and immunostaining. Briefly, cultures were blocked in blocking solution (5% normal goat serum and 0.3% TritonTM X-100 in PBS) for 2 h and then incubated overnight in primary antibody at 4°C. The following primary antibody was used: anti-βIII-tubulin (clone TUJ-1, 1:5000; R&D Systems no. 1195 V). Samples were gently washed three times with PBS and incubated with Alexa Fluor 647-conjugated goat anti-mouse in blocking solution (1:2000; ThermoFisher A21236) for 1 h at room temperature and finally washed three times in PBS. Images were captured with an Opera Phenix Scanner (PerkinElmer) using a ×20 objective.
Visualization of functional mitochondria
Live neuronal cultures were incubated with 50 nM TMRM (Image-iT™ TMRM Reagent I34361, ThermoFisher) for 45 min and imaged on the Opera Phenix using a 568 nm excitation laser. Total imaging time for one 96-well plate was 75 min. After TMRM imaging, cells were rinsed three times with PBS, fixed in 1% paraformaldehyde and immunostained for βIII-tubulin.
Sciatic nerve axotomy
Mice received doses as indicated (see figure legends and Supplementary material) by intraperitoneal injection with compound 4 in vehicle (10% DMSO/10% Solutol/80% saline), compounds 8 and 9 in vehicle (0.5% methyl cellulose, 0.1% Tween 80), or by oral gavage with compound 10 or vehicle (0.5% methyl cellulose, 0.1% Tween 80). After 30 min, mice were anaesthetized with isoflurane/O2. For sciatic nerve transection, animals received a skin incision at mid-thigh level and the sciatic nerve was exposed and traced back to the sciatic notch taking care to minimize damage to surrounding tissues. The nerve was cut 5 mm distal to the notch and a 1–2 mm piece was removed from the distal end to prevent reattachment of the cut ends. Finally, the skin was closed using fine surgical suture. Animals were kept warm until fully recovered. For 15-h experiments, animals treated with compounds 4, 8 and 9 received a second dose 8 h after the first one. Animals treated for 1 day received a third dose 10 h after the second dose. Animals treated with compound 10 received only a single dose. Mice received carpofen 5 mg/kg subcutaneous analgesia to minimize postoperative pain.
Plasma neurofilament light chain measurements
Fifteen hours after sciatic nerve axotomy (SNA), blood was collected via the saphenous vein and animals were killed by cervical dislocation or by CO2 inhalation, and blood was collected by cardiac puncture. Samples were placed on ice and centrifuged as early as possible for 5 min, at 10 000g and 4°C. Plasma, 15 µl, was transferred into 96-well round bottom plates for neurofilament light chain (NfL) determination. The remaining plasma was stored at −80°C. Samples were placed on ice and centrifuged as early as possible for 5 min, at 10 000g and 4°C. Plasma NfL was measured by enzyme-linked immunosorbent assay using the NF-Light enzyme-linked immunosorbent assay kit from Uman Diagnostics following the manufacturer’s instructions.
Metabolite extraction
Metabolites from sciatic nerves and cultured neurons were extracted as described previously.21 At time of tissue collection animals were sacrificed by cervical dislocation and sciatic nerves were quickly removed and placed on an ice-cold metal plate. The length of the individual nerve sample was measured using digital callipers, after removing the swollen end near the site of transection. Tissues were transferred to prefilled collection tubes (CK28 Precellys tube, Bertin Technologies) with 1 ml ice-cold acetonitrile (Carlo Erba) in water (Fischer Chemical) mixed with 1% formic acid (Acros organic) (3:1). Tubes were thawed on ice and homogenized with 10 µl of 2-chloroadenosine (2 ng/µl), used as an internal standard. The samples were ground three times at 4500 rpm for 90 s with a Precellys Evolution (Bertin Technologies). The homogenate was centrifuged at 4°C (4500g for 5 min) and 900 μl of the supernatant were transferred to a borosilicate tube. The sample was ground again in 1.1 ml of a solution of acetonitrile/water 1% formic acid (3:1) three times at 4500 rpm for 90 s. After centrifugation, 900 μl of the supernatant were transferred to the same borosilicate tube. The combined extracts were evaporated to dryness at 40°C in a Genevac EZ-2 Plus Evaporator and stored at −20°C until measurement. Mouse DRG neurons were prepared as described above and seeded at 100 000 cells/well as a 10 µl spotted culture in 24-well plates and placed in a humidified CO2 incubator for 15 min. After allowing for cells to adhere, culture media was gently added to a final well volume of 1 ml/well. Cells were treated at DIV6 and at the end of the experiment, tissue culture plates were placed on ice and culture medium replaced with ice-cold saline (0.9% NaCl in water, 500 μl per well). For collection of intracellular metabolites, saline was removed and replaced with 160 μl ice-cold 50% methanol in water. Cells were incubated for a minimum of 5 min on ice with the 50% methanol solution and then the solution was transferred to tubes containing 50 μl chloroform on ice, shaken vigorously and centrifuged at 20 000g for 15 min at 4°C. The clear aqueous phase (140 μl) was transferred into a microfuge tube and lyophilized under vacuum. Lyophilized samples were stored at −20°C until measurement. On the day of measurement, lyophilized samples were reconstituted with 15 µl of 5 mM ammonium formate (Sigma Millipore) and centrifuged at 15 000 rpm (20 000g) for 15 min at 4°C. Then 10 µl of clear supernatant was analysed by liquid chromatography coupled with mass spectrometry (LC-MS).
Metabolite measurement
Lyophilized samples were thawed on ice and suspended in 90 μl of water with 5 mM ammonium formate (Sigma Millipore). The homogenized solution was transferred in injection vials and mixed with 10 μl of internal standard (2-chloroadenosine at 2 ng/μl), of which 75 μl was used for analysis by online SPE coupled with liquid chromatography and tandem mass spectrometry (XLC-MS/MS). Supernatants from DRG neurons were stored frozen in 1.5-ml Eppendorf tubes, thawed on ice and 10 μl were transferred in an injection vial and mixed with 80 μl of water with 5 mM ammonium formate and 10 μl of internal standard (2-chloroadenosine at 2 ng/μl). Of this, 75 μl were used for analysis by online XLC-MS/MS. Online XLC-MS/MS samples were injected into an SPE cartridge (2 mm inside diameter, 1 cm length, packed with C18-HD stationary phase), part of the SPE platform from Spark Holland. Thereafter, the SPE cartridge was directly eluted on an Atlantis T3 column (3 μm, 2.1 × 150 mm; Waters) with a water/methanol with 5 mM ammonium formate gradient (0% B for 0.5 min, 0 to 40% B in 6 min, 40 to 60% B in 1 min, 60 to 100% B in 1 min, 100% B for 1 min and back to initial conditions), at a flow rate of 0.15 ml/min to the mass spectrometer. Metabolites (NAD, ADPR and cADPR) and the internal standard 2-chloroadenosine were analysed on a Quantiva triple quadrupole (Thermo Electron Corporation). Positive electrospray was performed on a Thermo IonMax electrospray ionization probe. To increase the sensitivity and specificity of the analysis, we worked in multiple reaction monitoring and followed the MS/MS transitions: NAD+ MH+, 664.1–136.1; ADPR MH+, 560.1–136.1; cADPR MH+, 542.1–136.1; 2Cl-Ade MH+, 302.1–170.1. The spray chamber settings were as follows: heated capillary, 325°C; vaporizer temperature, 40°C; spray voltage, 3500 V; sheath gas, 50 arbitrary units; auxiliary gas 10 arbitrary units. Calibration curves were produced by using synthetic NAD, ADPR, cADPR and 2-chloroadenosine (Sigma Millipore). The amounts of metabolites in the samples were determined by using inverse linear regression of standard curves. Values are expressed as nanograms per millimetre for sciaric nerves and nanograms per 100 000 cells for cultured neurons.
Paclitaxel CIPN model
On Days 1 and 2, animals (Sarm1 knockout mice, strain B6.129X1-Sarm1<tm1Aidi>/J; Jackson Laboratories, and/or wild-type C57BL/6J mice) were placed under heating lamps for 30 min. Mice then received either paclitaxel (50 mg/kg IV) or vehicle (12.5% Cremophor®, 12.5% ethanol: 75% saline) using intravenous catheter (29-gauge needle) via the tail vein. Dose volume (10 ml/kg). Animals were dosed orally by gavage with compound 10 in vehicle (0.5% methyl cellulose, 0.1% Tween 80) at 100 and 300 mg/kg daily starting 2 h before paclitaxel until the conclusion of the experiment. On Day 14, thresholds to mechanical stimulation applied to the hind paws were determined using von Frey filaments. Mice were habituated under Perspex boxes on top of a grid floor for at least 1 h beforehand. Observers performing the von Frey measurement were blind to experimental groups.
Nerve conduction studies
On Day 15 (SARM1 mutant study), or Days 9 and 15 (SARM1 inhibitor study), nerve conduction analysis was performed to measure tail nerve sensory nerve action potentials (SNAPs). Mice were anaesthetized with isoflurane/O2 and tail nerve conduction performed using a Viking Quest electromyography machine (Nicolet) by applying square wave pulses of 0.1 ms of duration (low filter set to 1 Hz and high filter to 10 Hz). Stainless steel electrodes were placed subcutaneously into the tail. Stimulating electrodes were placed distally and recording electrodes proximally, 30 mm from the stimulating electrode. A ground electrode was placed between the stimulating and recording electrodes. Intensity was increased until a maximal response was achieved, increased 20% further to obtain supramaximal stimulation and responses to trains of 20 stimuli were applied and mean amplitude and velocity was recorded. Amplitude was measured from baseline to first peak and conduction velocity calculated as a function of distal latency and distance between stimulating and recording electrode.
Immunohistochemistry and quantification of intraepidermal nerve fibre density
On Day 16 mice were euthanized by CO2 inhalation. Hind paw skin samples were removed and fixed in Zamboni’s fixative (Morphisto, no. 12773, pH 7.0). The plantar surface of each paw was removed, thoroughly rinsed in PBS, immersed in 30% sucrose for 48 h at room temperature and embedded in O.C.T. (Tissue Tec). Then, 50-µm thick longitudinal frozen sections were cut with a cryostat (Leica CM3050S) and collected in 24-well plates. A series of six sections was thoroughly rinsed in 1× PBS, permeabilized in 0.3% TritonTM X-100 in 1× PBS for 20 min and immersed in 10% normal goat serum for 30 min, Afterwards, sections were incubated in rabbit anti-Protein Gene Product 9.5 (1:250, Cedarlane, CL7756AP-50) for 72 h at room temperature with gentle agitation. Sections were thoroughly rinsed in PBS again and placed in CF488 goat antirabbit secondary antibody (Sigma) at a dilution of 1:1000 for 2 h at room temperature with gentle agitation. After further rinsing in 1× PBS, sections were counterstained with DAPI (Sigma-Aldrich, #D8417) to allow visualization of nuclei, mounted in 24-well SensoPlatesTM (Greiner) and covered with mounting medium (90% glycerol, 0.5% propyl gallate and 20 mM Tris, pH 9). The part of the plantar surface containing the footpad was identified and imaged on an Opera confocal microscope using a ×40 water objective and CCD camera with 1.3 megapixels. Intraepidermal nerve fibres (IENFs) crossing into the epidermis were determined in five fields of view per sample by examining the entirety of the 50-µm stack. Axons that crossed the basement membrane were counted, whereas secondary branching and epidermal nerve fragments that did not cross the basement membrane were not.22 The length of epidermis was measured at the level of the basement membrane and the density of IENFs (IENFs/mm) was obtained. IENF densities were averaged in the five fields of view for each animal. Imaging and analysis were done with the samples blinded to the observer.
Photomicrographs
Fluorescent images were obtained with an Opera confocal microscope using a ×40 water objective and CCD camera with 1.3 megapixels by taking a 30-plane z-stack. The z-stacks were reduced to one plane by using a maximum intensity projection algorithm with background correction.
Statistical analysis
Unless otherwise stated, data are reported as means ± standard error of the mean (SEM). All measurements were obtained from distinct samples. Statistical analyses were obtained with one-way or two-way ANOVA as indicated in the figure legends, with Holm–Sidak post hoc for comparisons between subgroups, as appropriate, P < 0.05 was considered statistically significant. Curve fitting and non-linear regression curves of pharmacological dose response experiments were calculated with a least-squares fit, log(inhibitor) versus response (four parameters). All graphs and statistics were drawn and calculated with GraphPad Prism software v.8.4. and details of the statistical analysis are provided in the respective figure legends.
Data availability
The datasets supporting the current study will be made available on reasonable request.
Results
Identification of isothiazole small molecule SARM1 NADase inhibitors
To identify novel small molecule inhibitors of SARM1 NADase activity, we used our previously described biochemical assay employing a construct containing the SAM and TIR domains of human SARM1 to measure production of ADPR, the hydrolysis product of NAD.19 A high-throughput screen of a collection of small molecule compounds resulted in the identification of the isothiazole compound 1 [half-maximal inhibitory concentration (IC50) of 4 µM; Fig. 1]. Subsequent optimization efforts were aimed at improving the potency of screening hit 1.
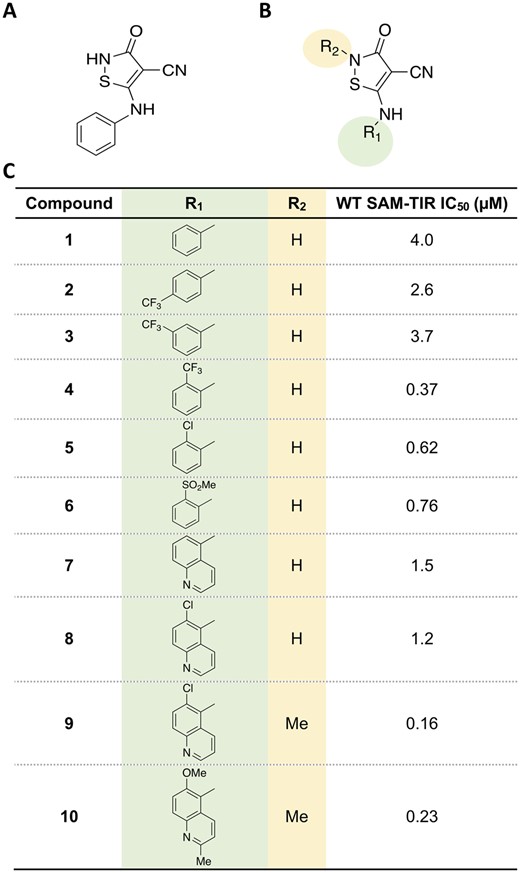
Identification of isothiazole inhibitors of SARM1 NADase. (A) Structure of isothiazole hit compound 1 identified as a weak inhibitor of ADPR production by NAD+ hydrolysis. (B) Core isothiazole structure subjected to structure-activity relationship studies. (C) Structure-activity relationship of the isothiazoles.
Structure-activity relationship expansion of isothiazole 1 evaluated modifications to the aryl group and explored impact of substitution on the isothiazole (Fig. 1). The ortho-CF3 aryl compound 4 provided an improvement in potency compared to 1 relative to the para (compound 2) and meta-(compound 3) isomers. The ortho-substituent could be replaced by both lipophilic and polar groups as well as bicycles without a detrimental impact on potency. Alkylation of the isothiazole core of 8 provided a robust improvement in potency in 9 that was coupled with further modifications to the isoquinoline ring system to provide 10, an orally available SARM1 inhibitor suitable for evaluation in chronic pharmacology models.
To assess selectivity for SARM1 NADase, the isothiazoles were tested for inhibition against a panel of other known NADases, PARPs, CD38, SIRTs, TNKSs and enzymes in the NAD pathway, NMNAT1 and NAMPT (Supplementary Table 1 and Supplementary Fig. 1).
Isothiazole small molecule inhibitors protect axons from Wallerian degeneration in vitro
To assess whether these pharmacological agents are capable of preventing Wallerian degeneration, we tested them in a cell-based assay using mouse DRG neurons in drop cultures and injured their axons by axotomy, as previously described.19,20 While axons from untreated neurons degenerated completely after injury, axons from DRG neurons treated with four isothiazole analogues, compounds 4, 8, 9 and 10, showed a dose-dependent resistance to axonal fragmentation caused by axotomy (Fig. 2A and B). The potency of the compounds in the cell assay tracked with their potency in the biochemical assay, in a manner consistent with their selectivity for SARM1 (e.g. 10 ≈ 4 ≈ 9 ≫ 8). In addition, we confirmed that isothiazoles also protected axons in axotomized human iPSC-derived motor neurons in a dose-dependent manner, with an efficacy and potency similar to mouse DRGs (Supplementary Fig. 2). To determine whether surviving cut axons were metabolically active, we exposed injured cultures to TMRM, which has been used to demonstrate the presence of functional mitochondria after injury in Sarm1−/− mutant neurons,23 and after pharmacological SARM1 inhibition.19 Untreated injured axons did not show TMRM fluorescence, indicating loss of viable mitochondria, whereas axons treated with compound 9 were both morphologically intact and preserved TMRM fluorescence in a manner qualitatively indistinguishable from uninjured axons (Supplementary Fig. 3). Together, these results demonstrate that isothiazole inhibitors of SARM1 NADase can prevent Wallerian degeneration of injured distal axons, similar to the recently described pharmacological inhibition of the NADase with reversible isoquinolines.19
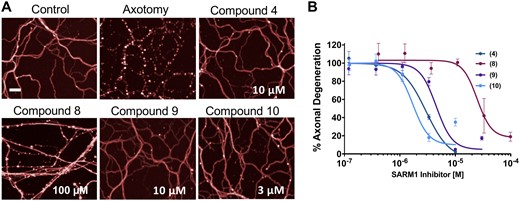
Isothiazole SARM1 inhibitors protect injured axons in vitro. (A) DRG mouse cultures were treated for 2 h with compounds 4, 8, 9 and 10 and then subjected to axotomy. Whereas axons in untreated cultures fragmented completely, axons from cultures treated with isothiazole inhibitors were completely protected 16 h post-axotomy. Scale bar = 25 µm. (B) Quantification of fragmentation showed that axonal protection with isothiazoles was dose-dependent. Values represent mean ± SEM n = 4/dose. Representative of three independent experiments with similar results.
Isothiazoles are irreversible SARM1 inhibitors
The isothiazole motif is present in a number of pharmacological agents that include kinase inhibitors,24 antivirals,25 herbicides and fungicides26,27 and others.28 Based on the reported mechanism of action of this class of compounds against other targets, which involves covalent modification of cysteine residues to form a disulphide adduct,29,30 we investigated the possibility that these may inhibit SARM1 through a similar mechanism. Washout experiments were performed with compounds 4 and 9 using plate-bound SAM-TIR to evaluate a covalent mechanism of action (Fig. 3A). Plate-bound SAM-TIR protein was incubated with the compounds for 2 h and rinsed. Enzymatic activity was assessed immediately, and at intervals of 10 and 60 min after removing the compounds. The IC50 of the isothiazoles was maintained unchanged 1 h after rinsing the plates, whereas the IC50 of the reversible isoquinoline SARM1 inhibitor DSRM-371619 was significantly right-shifted, showing an expected apparent loss of potency consistent with rapid release of compound bound to the target (Fig. 3A). In contrast, after isothiazole exposure, the complete maintenance of enzymatic inhibition after a 1 h washout interval is consistent with an irreversible mode of action, which was not affected by compound removal through rinsing of the plates. To ascertain the site of action of the isothiazoles, we conducted studies with mutants in the TIR domain. Within the enzymatic TIR domain of SARM1 there are three cysteine residues, C629, C635 and C649. Based on the TIR domain structure, C635 and C649 are exposed and may be amenable to modification with small molecules,31 thus, both were independently mutated to alanine. A subset of isothiazoles were evaluated against both the C635A SAM-TIR and C649A SAM-TIR mutants. Isothiazole compounds 4, 8, and 9 were significantly less potent in the C635A mutant compared to both wild-type and C649A SAM-TIR mutants, suggesting that a covalent modification may occur selectively at C635 (Table 1). Similar washout experiments were done with the C635A mutant and the IC50 did not shift after washout suggesting covalent modification can occur at C649 as well (data not shown).
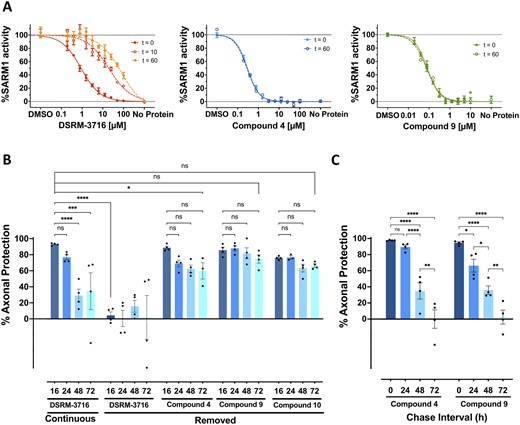
Isothiazoles are irreversible SARM1 inhibitors. (A) SAM-TIR protein immobilized to Strep-Tactin®XT plates was treated with DSRM-3716, compound 4 or compound 9 for 2 h and enzymatic activity was measured either immediately (t = 0), or plates were rinsed and assayed after 10 min (t = 10) or 60 min (t = 60) post removal of the compounds. Substantial loss of inhibition was observed with the reversible SARM1 inhibitor DSRM-3716 (left). In contrast, inhibition with compound 4 (middle) and compound 9 (right) was completely maintained after 60 min in the absence of compound, consistent with irreversible inhibition of the enzyme. (B) DRG mouse cultures were treated for 3 h with 10 µM of the reversible SARM1 inhibitor DSRM-3716 or 10 µM each of compounds 4, 9 and 10. Compounds were removed from the cultures and a subset of cultures had DSRM-3716 replaced immediately after rinsing (continuous). Axons were subjected to axotomy and examined at the times indicated below the bars (in hours). Whereas protection by DSRM-3716 was completely lost by compound removal at the 16-h time point, compounds, 4, 9 and 10 maintained axonal protection at 72 h. ANOVA with Holm–Sidak post hoc F(11,36) = 10.49, P < 0.0001, mean ± SEM; control n = 4. ns, not significant; **P < 0.01; ****P < 0.0001. (C) DRG mouse cultures were exposed to a 3-h pulse of 10-µM compound 4 or 10-µM compound 9 and compounds were removed from the cultures. The interval between removal of the compound and axotomy defines the chase interval. At the completion of the chase interval indicated below the bars, axons were subjected to axotomy and axonal protection was examined after 16 h. Almost complete axonal protection was maintained after a chase period of 24 h. The extent of axonal protection progressively decreased with longer intervals between compound removal and axotomy. Two-way ANOVA with Holm–Sidak post hoc F(3,24) = 71.81, P < 0.0001, mean ± SEM; n = 4. *P < 0.05; **P < 0.01; ****P < 0.0001.
Loss of biochemical potency against C635A SARM1 mutants, combined with the previously described covalent mode of action by other isothiazole drugs,29,30 led us to explore whether axonal protection by this class of inhibitors could occur by irreversible inactivation of SARM1. To test this hypothesis, we exposed DRG neurons to a 3 h pulse of SARM1 inhibitor compound 4, as well as compound 9 and its close analogue compound 10, each at 10 µM a dose that provides maximum protection from axotomy, removed the compound from the cultures and subsequently injured the cells immediately after the inhibitor was removed. Consistent with a mechanism involving irreversible inactivation of the enzyme, injured axons exposed to isothiazole SARM1 inhibitor before injury showed prolonged protection, up to 72 h post-axotomy, without any evidence of axonal damage due to SARM1 reactivation due to removal of the compound (Fig. 3B). In contrast, the previously described reversible isoquinoline DSRM-371619 did not provide any protection when axons were injured immediately after compound removal (Fig. 3B). These results also indicate that the extent of axonal protection with isothiazoles in vitro is similar to Sarm1 genetic loss-of-function.
In this experimental paradigm, where axons were severed from their respective somas, there cannot be SARM1 resynthesis and resupply from the cell body. The long-lasting protection observed after compound removal suggested that the inhibitor acted as a stable covalent inhibitor of pre-existing SARM1 protein contained in the axons before injury. We took advantage of this property to explore, in intact neurons, how long would it take to replenish SARM1 to levels required to restore an axonal fragmentation response, after irreversible inhibition of pre-existing enzyme and compound removal. To test this hypothesis, we exposed DRG neurons to a 3-h pulse of compounds 4 and 9 at 10 µM that, because of their similar potency, provides maximal protection from axotomy for both compounds. When axotomy was progressively delayed after removing the compound, the SARM1-dependent degenerative response was also progressively restored, until it was completely re-established after 72 h. This is consistent with sufficient protein resynthesis during that 72-h window to allow re-establishment of the SARM1-dependent local injury response (Fig. 3C).
SARM1 inhibitors prevent cADPR increase and NfL release from axotomized nerves in vivo
Numerous literature reports have described that loss of SARM1 provides axon protection following axotomy in vivo.32–34 We recently reported that axonal degeneration after SNA can be monitored 15 h post-injury by measuring plasma levels of NfL and increases in cADPR within injured nerves, both occurring in a SARM1-dependent manner.21 NfL is an axonal cytoskeletal protein released into circulation during traumatic axonal degeneration, and cADPR is a direct product of SARM1 NADase activity.21 To rapidly identify inhibitors of SARM1 that could be advanced for testing in a chronic model of neuropathy, we evaluated selected compounds from the isothiazole series for their ability to prevent increased NfL released into circulation by severed sciatic nerves at 15 h post-transection. Mean baseline plasma NfL levels from six independent cohorts were 65.0 ± 3.8 pg/ml (mean ± SEM). In contrast, mean plasma NfL levels 15 h after SNA in 14 independent cohorts resulted in an increase to 2395.1 ± 74.2 pg/ml (mean ± SEM), i.e. a ∼37-fold increase from baseline (Supplementary Fig. 4B). Since the range of mean plasma NfL levels in these 15-h injured cohorts varied ∼2-fold (1781–3614 pg/ml) comparison of treatment effects and effect sizes between independent experiments may require normalization rather than absolute values. Compounds 4, 9 and 10 were well-tolerated at doses that detectably reduced plasma NfL in a dose-dependent manner 15 h after sciatic nerve injury (Fig. 4A). All three compounds showed similar efficacy in vivo and reduced NfL levels by ∼60% at the maximum tolerated dose; however, only compound 10 was compatible with oral dosing. We also confirmed that these compounds reduced cADPR produced in injured nerves, in a manner consistent with inhibition of SARM1 NADase (Supplementary Fig. 4A).
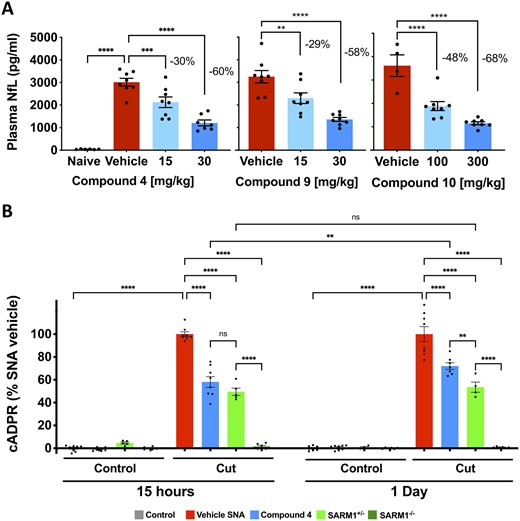
Isothiazoles inhibit SARM1 in vivo in SNA. (A) Mice were treated with isothiazole SARM1 inhibitors 4, 9 and 10 at the doses indicated and subjected to unilateral SNA 30 min after the first dose. For compounds 4 and 9, a second dose was administered 8 h after the first dose, and 15 h after SNA NfL levels were measured in plasma. Compounds 4 and 9 were dosed by intraperitoneal injection and compound 10 was dosed once orally. All three compounds prevented increases in plasma NfL in a dose-dependent manner. The magnitude of the effect, expressed as percentage decrease from vehicle is as indicated in the figures. Values represent mean ± SEM. Compound 4, ANOVA with Holm–Sidak post hoc F(2,20) = 22.48, P < 0.0001, n = 7–8/group; compound 9, ANOVA with Holm–Sidak post hoc F(2,21) = 19.38, P < 0.0001, n = 8 per group; compound 10, ANOVA with Holm–Sidak post hoc F(2,17) = 28.54, P < 0.0001, vehicle n = 4, treated groups n = 8. **P < 0.01; ****P < 0.0001. (B) Wild-type, Sarm1+/− and Sarm1−/− mice were subject to SNA. At 15 h and 1 day after axotomy cADPR was measured in the cut and uncut contralateral nerves. Wild-type mice were treated with compound 4 (30 mg/kg) 30 min before SNA and received a second dose 8 h after the first dose in the 15-h group, and a third dose 10 h after the second dose in the 1-day group (TID dosing). We found cADPR increased in cut nerves but not the contralateral uncut nerves in wild-type. In Sarm1 mutants, no cADPR increase was observed in Sarm1−/− at any time point and values were intermediate in Sarm1+/−. Levels of cADPR with compound 4 were reduced from vehicle to levels that approached Sarm1+/−. Values were normalized to the respective SNA vehicle within each group and represent mean ± SEM. One-way ANOVA F(15,88) = 188.2, P < 0.00001; ns. not significant; **P < 0.01; ****P < 0.0001; n = 4–8/group.
To estimate the extent and duration of SARM1 inhibition with this chemical series, we used treatment with compound 4 as an example to compare the magnitude of reduction of the proximal SARM1 biomarker cADPR in injured wild type mice, with that observed in Sarm1+/− and Sarm1−/− at 15 h and 1 day after nerve transection (Fig. 4B). As expected, cADPR production was inhibited completely in Sarm1−/−. At 15 h, compound 4 (30 mg/kg) inhibited cADPR to a similar extent to Sarm1+/− (compound 4 = 58.1 ± 5.7%; Sarm1+/− = 49.5 ± 3.2%). One day after nerve transection, cADPR levels with compound 4 had increased significantly, to 72% of control, while they were still 53.5% in Sarm1+/−. These observations suggest that SARM1 inhibition with the maximum tolerated dose of compound 4 approached the level of SARM1 activity present in Sarm1+/−.
SARM1 inhibitors protect axons and partially prevent development of CIPN induced by paclitaxel
SARM1 genetic loss-of-function has been previously shown to protect axons and prevent the appearance of peripheral neuropathy in preclinical models of CIPN using vincristine,9 paclitaxel14 or bortezomib.10 To define the parameters needed to generate a SARM1-dependent neuropathy in C57BL/6 mice, we treated wild-type and Sarm1−/− mutant mice with paclitaxel and measured the effect on SNAP amplitude and conduction velocity. We determined that two doses of 50 mg/kg paclitaxel induced a profound neuropathy characterized by a 65% decrease in the SNAP amplitude of the tail nerve (Supplementary Fig. 5). This is consistent with a neuropathy driven primarily by decreases in the number of viable axons, as has been observed previously.9,14 When we examined the effect of paclitaxel in Sarm1 mutant mice, we noticed robust protection of SNAP amplitudes in tail nerves from Sarm1−/−. Furthermore, this protective effect was gene-dosage dependent and Sarm1 heterozygous mice showed partial preservation of SNAP amplitudes at values that were intermediate between wild-type mice and homozygous Sarm1 knockout mice (Supplementary Fig. 5).
The protection of tail nerve SNAP amplitudes provided by Sarm1 genetic loss-of-function suggested that an inhibitor of the SARM1 NADase would have the potential to treat CIPN. Therefore, we tested the translational potential of pharmacological SARM1 inhibition with the orally bioavailable isothiazole compound 10, in the mouse paclitaxel CIPN model. Animals exposed to two doses of 50 mg/kg paclitaxel showed a decrease in tail nerve SNAP amplitudes 9 days after the first dose of paclitaxel, which remained at similarly low levels 2 weeks after the first dose of paclitaxel (Fig. 5A). Animals exposed to paclitaxel also experienced loss of IENFs (Fig. 5D and E), and decreased threshold for mechanical stimulation assessed by von Frey filaments (Fig. 5C) after 2 weeks. We observed that vehicle-treated animals presented an increase in SNAP amplitude proportional with age, similar to a normal developmental increase in tail SNAP amplitudes reported in rats.35–37 Paclitaxel-treated animals that received oral administration of the SARM1 inhibitor compound 10, exhibited partial preservation of SNAP amplitudes during the first and second weeks post-paclitaxel at 300 mg/kg but not at 100 mg/kg (Fig. 5A). Consistent with the preservation of axonal function in tail nerves, we also observed partial preservation of the threshold for mechanical stimulation (Fig. 5C) and complete preservation of small calibre axonal structures in IENFs (Fig. 5D and E). Together, these results indicate that treatment with an orally bioavailable small molecule inhibitor of SARM1 NADase activity protected axonal integrity and function from neuropathy induced by paclitaxel. We compared the extent of protection of SNAP amplitudes obtained with compound 10 with that observed in Sarm1 heterozygous and homozygous mutants (Supplementary Fig. 5). After normalization to each respective vehicle control group, the percentage protection of SNAP amplitudes obtained with the high dose of compound 10 was 44.7 ± 12.9% at 9 days and 23.1 ± 7.3% at 15 days post-paclitaxel (Fig. 5F; mean ± SEM). In contrast, in Sarm1 mutants, the protection of SNAP amplitude at 15 days post-paclitaxel was 33.2 ± 5.2% in Sarm1+/− and 80.4 ± 6.4% in Sarm1−/− (Fig. 5F; mean ± SEM). This analysis indicates that compound 10 protected SNAP amplitudes to an extent similar to the protection afforded by Sarm1 heterozygous mutants.
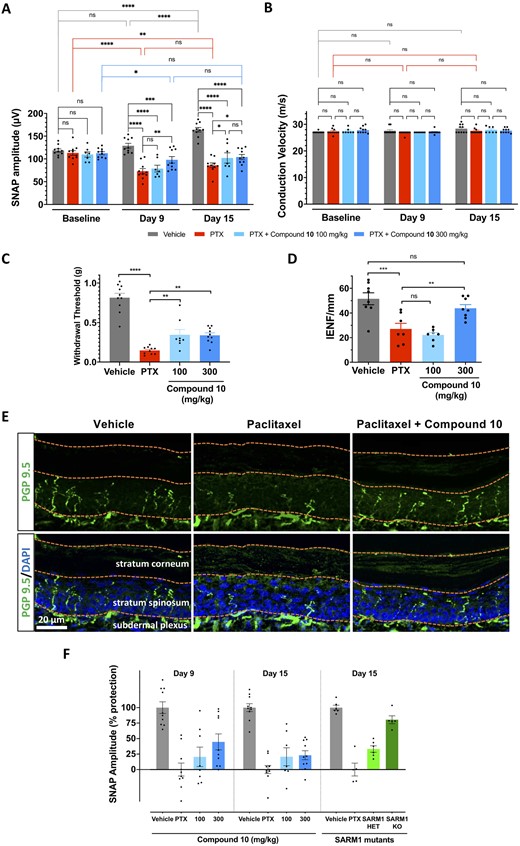
Pharmacological SARM1 inhibitor protects axonal integrity and function in a model of CIPN. Mice were subjected to the paclitaxel CIPN model described in the 'Materials and methods' section and Supplementary Fig. 5, while treated orally with vehicle, or the isothiazole SARM1 inhibitor compound 10 at the doses indicated in the figure. (A) SNAP amplitudes from tail nerves measured at 9 and 15 days after the first dose of paclitaxel were partially protected at the highest dose, while also showing a non-statistically significant trend towards protection at the lower dose. There was a significant main effect between groups, two-way repeated measures ANOVA F(3,33) = 28.12, P < 0.0001; time F(2,66) = 17.37, P < 0.0001; Time × Treatment group F(6,66) = 10.61, P < 0.0001; Holm–Sidak post hoc *P < 0.05; **P < 0.01; ****P < 0.0001; n = 7–10 per group. (B) Nerve conduction velocity was not affected by paclitaxel or compound treatment. There was no significant main effect by two-way ANOVA, treatment groups F(3,33) = 1.287, P = 0.2951 and Time × Treatment group F(6,66) = 1.146, P = 0.346; n = 7–10 per group. (C) Mechanical withdrawal threshold was significantly reduced by paclitaxel treatment and showed partial protection by treatment with compound 10. One-way ANOVA F(3,34) = 43.43, P < 0.0001, Holm–Sidak post hoc **P < 0.01; ****P < 0.0001; n = 7–10 per group. (D) IENFs were stained with the pan-axonal antibody PGP 9.5 and confocal microscopy images were quantified by a blind experimenter as described in the 'Materials and methods' section. Loss of IENF density induced by paclitaxel was significantly protected by SARM1 inhibitor. One-way ANOVA F(3,26) = 13.16, P < 0.0001; Holm–Sidak post hoc **P = 0.0091; ***P = 0.0003; n = 7–8 per group. (E) Representative images of IENFs in vehicle, paclitaxel and paclitaxel + 300 mg/kg compound 10. (F) Percentage protection of SNAP amplitudes in mice treated with paclitaxel and compound 10 at 9 and 15 days versus percentage protection achieved through genetic reduction of SARM1 in Sarm1 heterozygous (HET) and Sarm1 knockout (KO) at 15 days. The magnitude of protection with the highest dose of compound 10 approached that of Sarm1 heterozygous.
Discussion
SARM1 is the central mediator of a cell autonomous programme to dismantle axons in response to a variety of pathological insults that include mechanical or traumatic injuries, mitochondrial dysfunction, metabolic syndrome and chemotherapeutic agents.4,5,7 These insults have been implicated in a pathological dying-back process of neurodegeneration that begins in the axon and is common to many neurological conditions. Many attempts at neuroprotection targeting the cell body of the neuron have been unsuccessful in the clinic.38 This has led to an increased recognition of the importance of the early and ongoing damage to the axon in the overall pathophysiology and long-term outcome of neurological conditions involving axonopathy as a main driver of disease. SARM1 is uniquely positioned as a target for therapeutic intervention for these neurodegenerative conditions because of its central role as the mediator of axonal degeneration and also because its mechanism of action involves an NADase enzymatic activity amenable to pharmacological intervention.3,4
SARM1 can be inhibited irreversibly by small molecules that reproduce SARM1 loss-of-function
Recently, we demonstrated that pharmacological inhibition of the SARM1 NADase with small molecules was possible with a series of reversible isoquinolines. Those compounds were capable of protecting injured axons in vitro and allowed recovery of axons from a metastable condition induced by mitochondrial dysfunction.19 To extend these findings and evaluate SARM1 pharmacological inhibition in a preclinical model of axonopathy, we identified a new isothiazole chemical series of SARM1 NADase inhibitors, and developed an orally bioavailable compound suitable for chronic dosing in vivo. A series progression and optimization led to selected small molecules that showed submicromolar potency against recombinant SARM1, and good specificity against other NADases and a commercial panel of enzymes and kinases. Three potent members of this series exhibited partial inhibition of NMNAT1 at 10 µM in the cell-free assay (Supplementary Table 1). It is plausible that a similar interaction occurs with the closely-related isoform NMNAT2, the main known negative regulator of SARM1. However, with an IC50 for SARM1 ranging from 0.16 µM to 0.37 µM, these inhibitors have a >30-fold greater potency for SARM1 inhibition over NMNAT1. In addition, we confirmed with compound 10 that there were no changes in NAD+ in neuronal cultures treated at doses that provide maximal axonal protection. Therefore, it is unlikely that there was substantial inhibition of NMNATs in our assays. Treatment of cultured mouse DRG neurons and human iPSC-derived motor neurons with these isothiazoles showed robust dose-dependent protection of severed distal axons, which approached the level and duration of protection observed in Sarm1 homozygous null. Three independent sets of experimental observations support that isothiazole SARM1 inhibitors act as irreversible inhibitors of SARM1 and that the irreversible inhibition may occur through interaction with an exposed cysteine. The first is the ability of isothiazoles to maintain SARM1 in an inhibited state in a plate-bound biochemical assay after compound removal, while the same experiment using the previously described reversible inhibitor DSRM-3716 allowed SARM1 to progressively recover enzymatic function. The second is the change in potency observed when isothioazoles were tested on SARM1 constructs where Cys635 was mutated to an alanine. The C635A mutants are enzymatically active and can use NAD+ as a substrate. Nevertheless, isothiazoles showed substantially reduced potency against C635A mutant constructs, compared to wild-type or to C639A mutants. The third experimental piece of evidence supporting irreversible inactivation was obtained in cells. Since axotomized neurons do not resupply SARM1 into the distal axon, irreversible inactivation would be expected to provide sustained axonal protection in the absence of the continuous presence of an inhibitor. We observed that a brief exposure of neurons to isothiazoles, before injury, was sufficient to confer long-lasting protection to injured axons after the compound was removed from the cells. The extent and duration of this protective effect in vitro was similar to what has been reported for Sarm1 knockout neurons32,33 indicating that pharmacological SARM1 inhibition with these isothiazoles can approach the levels provided by complete genetic loss-of-function. However, if excess, unbound irreversible inhibitor is removed from intact neurons before injury, protein synthesis would be expected to eventually resupply axons with sufficient de novo enzyme to restore their ability to undergo Wallerian degeneration after axotomy. We obtained direct experimental evidence that this was indeed the case. As the interval between removal of the compound and axotomy was extended, neurons displayed increased recovery of their ability to undergo Wallerian degeneration after axotomy. We interpret the combined observations in the biochemical and cell-based experiments as evidence of irreversible enzymatic inactivation by the isothiazoles. This may be due to covalent modification of C635 that resides in the SS loop, which leads to structural changes that lock the C-terminus in an inactive conformation.
SARM1 inhibitors prevent increases in nerve cADPR and plasma NfL released by traumatic injury in vivo
To test SARM1 inhibitors in vivo we first needed to evaluate and optimize the pharmacokinetic (PK) properties of the isothiazoles to make them suitable for dosing and then assess modulation of SARM1. We decided to assess the pharmacodynamic (PD) properties of candidate isothiazoles in modulating the levels of cADPR within the nerve and plasma NfL released into circulation after nerve transection. cADPR is a biomarker directly produced by SARM1 NADase21 and NfL is released into circulation after traumatic axonal damage. We have previously demonstrated that, in sciatic nerve axotomy, increases in both biomarkers are SARM1-dependent and can be detected as early as 15 h post-transection of sciatic nerves.21 Using an SNA model of axonal injury in vivo, we determined that three isothiazole SARM1 inhibitors were similarly able to reduce release of plasma NfL by ∼60% and also reduced cADPR. Using compound 4 to monitor changes in cADPR we also estimated that the efficacy of these compounds in vivo was inferior to Sarm1−/− and, unlike their robust efficacy displayed in vitro, approached that of Sarm1 heterozygosity. We hypothesize that this discrepancy between in vitro and in vivo efficacy is likely due to limitations of drug tolerability, which prevented the use of higher dosing.
Paclitaxel CIPN in mouse is a clinically relevant model of human neuropathy
To test the concept that pharmacological SARM1 inhibition could protect axons in peripheral neuropathies, we chose a model of paclitaxel-induced CIPN. Taxanes represent an important class of widely used chemotherapeutic agents that are known to cause neuropathy. Previous studies had shown that nerve damage induced by paclitaxel and other chemotherapeutic agents was substantially reduced in WldS39 and Sarm1 knockout mice,9,10,14 suggesting that activation of the Wallerian-like degeneration pathway triggered by SARM1 is mechanistically responsible for that neuropathy. We implemented and characterized a paclitaxel preclinical model in mice that induces a SARM1-dependent neuropathy at doses that have clinical equivalence,40,41 and determined the parameters required to test pharmacological candidates. Although this model is substantially shorter than the most common clinical course of paclitaxel, which typically takes weekly infusions over 12 weeks, it still recapitulates most, if not all, of the hallmark neuropathological symptoms induced in the clinic. These include damage to long myelinated sensory fibres, IENF and tactile sensitivity.14,39 Indeed, we confirmed in our model that paclitaxel induced a severe neuropathy and Sarm1 genetic deletion provided robust protection of axonal function and integrity assessed by preservation of SNAP amplitudes.
Furthermore, and somewhat surprisingly, we also observed a gene-dosage effect of SARM1 protection, with the values of SNAP amplitudes in heterozygous mice treated with paclitaxel falling between those of wild-type and Sarm1 knockout mice. This finding is significant because, until recently, literature reports using axotomized animals had shown complete axonal protection of distal nerves in Sarm1 homozygous mutants, while heterozygous animals were assumed not to have a protective phenotype and were used as controls.32,42 The gene-dosage effect in paclitaxel-induced CIPN is relevant to therapy because in this model of Wallerian-like degeneration the intermediate level of protection it provides is long-lasting. We attribute this difference between SNA and paclitaxel to the different nature of the insult. Whereas in axotomy the injury is immediate, complete and catastrophic, the nerve injury after paclitaxel treatment develops more slowly and occurs in neurons that are initially intact. In severe and extreme injuries, such as complete nerve transection or crush, Sarm1 heterozygosity is unable to provide long-lasting axonal protection.32 It is likely, in light of our results, that paclitaxel induces a slower evolving and transient injury, thus allowing partial SARM1 loss-of-function to provide sufficient protection, compared to axons that have been injured by trauma. Consistent with this interpretation, a long-lasting gene-dosage protective effect of SARM1 loss-of-function was recently reported in a chronic model of photoreceptor degeneration that takes several weeks to develop.43 Therefore, it is plausible that a protective effect from partial inhibition of SARM1 may still allow for a therapeutic benefit when using pharmacological agents.
After confirming adequate PK/PD properties in the SNA model, we tested compound 10, an orally bioavailable isothiazole SARM1 inhibitor, in the mouse paclitaxel CIPN model. We found that pharmacological inhibition of SARM1 protected axonal structure, as evidenced by robust maintenance of IENFs in foot pads, as well as partial functional protection in recordings of SNAP amplitudes and reduction in mechanical sensitivity thresholds evaluated by von Frey filaments. Similar to the partial efficacy observed in SNA, the efficacy obtained in the paclitaxel CIPN model approached that achieved by Sarm1 heterozygous mutants rather than Sarm1 null. Nevertheless, even with those limitations, these compounds provided a novel tool to test and demonstrate the translational hypothesis that pharmacological inhibition of SARM1 NADase can be therapeutically beneficial in CIPN. While compound 10 provided robust protection of small calibre axons in IENF, protection of large calibre and long myelinated axons was more limited. This may reflect different sensitivity of these different fibres to paclitaxel or to SARM1 activation. In a recent study using paclitaxel in Sarm1−/−, Turkiew et al.14 also observed an apparently increased sensitivity of long myelinated fibres to paclitaxel, although our current observations indicate that loss of SNAP amplitudes in the tail nerve is indeed a SARM1-dependent process. Consistent with the protective effect of Sarm1 heterozygotes, our data with compound 10 suggest that partial pharmacological inhibition of SARM1 may provide substantial and clinically relevant therapeutic benefit in CIPN. Although we obtained almost complete protection of IENF, the partial protection of SNAP amplitudes and tactile allodynia indicate that there is a window for improvement, and it is likely that to obtain the full benefit of SARM1 loss-of-function, pharmacological agents will need to achieve higher levels of inhibition in vivo.
Therapeutic applications of SARM1 inhibition
From a therapeutic translational perspective, CIPN represents a unique clinical paradigm, with substantial unmet medical need for an effective therapy16 and a predictable clinical outcome that lends itself to test axonal protection therapies. Using this paradigm, we provided the first demonstration that SARM1 inhibition with pharmacological agents that target the NADase can provide a therapeutic benefit in an animal model of human neuropathy. The small molecule inhibitors described in this study are research tools that allowed execution of this proof of principle preclinical study. Although here we used a new class of irreversible inhibitors, we recently described the development of potent reversible small molecule SARM1 inhibitors.19 At the moment it is not clear which mode of binding is superior to achieve the goal of high and sustained inhibition.
Peripheral neuropathies are the most common neurodegenerative condition, affecting millions of patients. Beyond CIPN, diabetic peripheral neuropathy is also a candidate for treatment using SARM1 inhibitors, as evidenced by protection mediated by loss of Sarm1 in preclinical models.8,14 Amongst the inherited neuropathies, some models of Charcot-Marie-Tooth disease have shown protection in the WldS background,11,13 which acts via chronic SARM1 inhibition to provide protection from Wallerian and Wallerian-like degeneration.34
The role of SARM1 and Wallerian-like degeneration in the CNS has also been demonstrated in models of traumatic brain injury44–46 and glaucoma47 and the potential applications of SARM1 inhibition in chronic CNS degenerative conditions is the subject of active research.4,5 We anticipate that the development and availability of pharmacological reagents to inhibit SARM1 in vivo will allow therapeutic testing in additional models of peripheral and central axonopathies.
Acknowledgements
We thank Aaron DiAntonio, Jeff Milbrandt and members of Disarm Therapeutics for critical reading of the paper. We also thank Markus Ketelhot, Hanno Ewers, Daniel Küver, Michael Grün, Johanna Nolte, Nico Bürger, Malik Majandinow, Nicole Hoeschen, Esther Strerath, Christin Winkler, Benjamin Morisse and Niels Banek for invaluable technical help in the course of these studies.
Funding
This study was funded by Disarm Therapeutics.
Competing interests
T.B., R.O.H, T.M.E., R.D. and R.K. are employees and shareholders of Disarm Therapeutics, a wholly owned subsidiary of Eli Lilly & Co., and inventors on patents related to this work. The authors have no other competing conflicts or financial interests.
Supplementary materials
Supplementary material is available at Brain online.
References
- CIPN
chemotherapy-induced peripheral neuropathy
- DRG
dorsal root ganglion
- IENF
intraepidermal nerve fibre
- NfL
neurofilament light chain
- SNA
sciatic nerve axotomy
- SNAP
sensory nerve action potential


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}