




Abstract
We describe a recently recognized disease entity, limbic-predominant age-related TDP-43 encephalopathy (LATE). LATE neuropathological change (LATE-NC) is defined by a stereotypical TDP-43 proteinopathy in older adults, with or without coexisting hippocampal sclerosis pathology. LATE-NC is a common TDP-43 proteinopathy, associated with an amnestic dementia syndrome that mimicked Alzheimer’s-type dementia in retrospective autopsy studies. LATE is distinguished from frontotemporal lobar degeneration with TDP-43 pathology based on its epidemiology (LATE generally affects older subjects), and relatively restricted neuroanatomical distribution of TDP-43 proteinopathy. In community-based autopsy cohorts, ∼25% of brains had sufficient burden of LATE-NC to be associated with discernible cognitive impairment. Many subjects with LATE-NC have comorbid brain pathologies, often including amyloid-β plaques and tauopathy. Given that the ‘oldest-old’ are at greatest risk for LATE-NC, and subjects of advanced age constitute a rapidly growing demographic group in many countries, LATE has an expanding but under-recognized impact on public health. For these reasons, a working group was convened to develop diagnostic criteria for LATE, aiming both to stimulate research and to promote awareness of this pathway to dementia. We report consensus-based recommendations including guidelines for diagnosis and staging of LATE-NC. For routine autopsy workup of LATE-NC, an anatomically-based preliminary staging scheme is proposed with TDP-43 immunohistochemistry on tissue from three brain areas, reflecting a hierarchical pattern of brain involvement: amygdala, hippocampus, and middle frontal gyrus. LATE-NC appears to affect the medial temporal lobe structures preferentially, but other areas also are impacted. Neuroimaging studies demonstrated that subjects with LATE-NC also had atrophy in the medial temporal lobes, frontal cortex, and other brain regions. Genetic studies have thus far indicated five genes with risk alleles for LATE-NC: GRN, TMEM106B, ABCC9, KCNMB2, and APOE. The discovery of these genetic risk variants indicate that LATE shares pathogenetic mechanisms with both frontotemporal lobar degeneration and Alzheimer’s disease, but also suggests disease-specific underlying mechanisms. Large gaps remain in our understanding of LATE. For advances in prevention, diagnosis, and treatment, there is an urgent need for research focused on LATE, including in vitro and animal models. An obstacle to clinical progress is lack of diagnostic tools, such as biofluid or neuroimaging biomarkers, for ante-mortem detection of LATE. Development of a disease biomarker would augment observational studies seeking to further define the risk factors, natural history, and clinical features of LATE, as well as eventual subject recruitment for targeted therapies in clinical trials.
Introduction
Transactive response DNA binding protein of 43 kDa (TDP-43) proteinopathy in limbic brain structures is commonly observed in subjects past 80 years of age. This proteinopathy has been associated with substantial cognitive impairment that mimicked Alzheimer’s disease clinical syndrome in retrospective studies. Despite evidence from many sources attesting to the public health impact of age-related TDP-43 proteinopathy, there is as yet no consensus-based nomenclature. To address this problem, we propose new terminology: limbic-predominant age-related TDP-43 encephalopathy (LATE). Guidelines are suggested for the autopsy evaluation and staging of LATE neuropathological change (LATE-NC). We review the medical literature pertaining to LATE, including cognitive manifestations, neuroimaging, public health impact, and genetics. The importance of LATE as a contributing factor in neurodegeneration is stressed, as are the needs for specific LATE biomarker development, TDP-43 focused drug discovery, and eventual clinical trials. We conclude by highlighting important knowledge gaps and potential future directions for research on LATE. Summary points are presented in Box 1.
LATE and LATE-NC summary points
LATE-NC features
A sampling and staging system for routine autopsy diagnosis is proposed to characterize the anatomical distribution of TDP-43 proteinopathy
Stage 1: amygdala only
Stage 2: +hippocampus
Stage 3: +middle frontal gyrus
Hippocampal sclerosis pathology may be observed (and should be reported), but is neither necessary nor sufficient for diagnosis of LATE-NC
LATE-NC is present in >20% (up to 50%) of individuals past age 80 years according to large community-based autopsy series
LATE is associated with substantial disease-specific cognitive impairment, usually an amnestic dementia syndrome (‘dementia of the Alzheimer’s type’)
The overall public health impact of LATE is on the same order of magnitude as Alzheimer’s disease neuropathological changes; the diseases are often comorbid, but which pathology is more severe varies greatly between individuals
Genetic risk factors for LATE have some overlap with FTLD-TDP and with Alzheimer’s disease
There is no molecule-specific biomarker for LATE. This is an important area of need for use in clinical trials (including as a potential exclusion criterion for Alzheimer’s disease clinical trials) and longitudinal studies of the clinical and pathological progression of LATE
Background
There is growing awareness that Alzheimer’s disease neuropathological change (ADNC) is only one of multiple neuropathological substrates associated with amnestic mild cognitive impairment and the Alzheimer’s clinical syndrome in the aged population (Korczyn, 2002; Zekry et al., 2002; Bennett et al., 2006; Jellinger and Attems, 2007; Schneider et al., 2007; Crary et al., 2014; Murray et al., 2014; Rahimi and Kovacs, 2014; James et al., 2016). Recent studies have gathered rich clinical data from large groups of subjects across a spectrum of cognitive states, correlated these clinical findings with new pathological markers at autopsy, and then analysed the data using powerful statistical methods. These studies have indicated that the diseases of aged human brains are complex: multiple comorbid pathologies are the norm, and there is substantial interindividual variation in neuropathological phenotypes (Neuropathology Group. Medical Research Council Cognitive and Aging, 2001; Brayne et al., 2009; Kovacs et al., 2013; Murray et al., 2014; Rahimi and Kovacs, 2014; White et al., 2016; Abner et al., 2017; Kapasi et al., 2017; Suemoto et al., 2017; Tanskanen et al., 2017; Robinson et al., 2018b, c). While there is a strong association between severe ADNC and cognitive impairment in all age groups (Nelson et al., 2009; Abner et al., 2011), subjects who die after 80 years of age often have exhibited cognitive decline exceeding expectation given the severity of ADNC (Kawas and Corrada, 2006; Savva et al., 2009; Nelson et al., 2012). LATE-NC is an important contributor to this apparent clinicopathological mismatch (see below).
Historically, the first-recognized pathological manifestation of LATE was profound hippocampal neuron loss and gliosis, collectively termed hippocampal sclerosis. In a landmark study, Dickson et al. (1994) identified 13 elderly subjects with dementia and hippocampal sclerosis, yet who lacked substantial ADNC. Other larger autopsy series that included subjects with dementia and hippocampal sclerosis were later reported (Crystal et al., 2000; Barker et al., 2002; Leverenz et al., 2002; White et al., 2002; Zarow et al., 2005; Attems and Jellinger, 2006; Brayne et al., 2009). In 2006, phosphorylated TDP-43 was discovered as the disease protein in the ubiquitylated inclusions that are signatures of amyotrophic lateral sclerosis (ALS) and most cases of frontotemporal lobar degeneration (FTLD), known as FTLD-TDP (Neumann et al., 2006; Cairns et al., 2007a). TDP-43 protein, encoded by the TARDBP gene (Ou et al., 1995), is a protein that binds to RNA and DNA as well as to other proteins, and serves multiple functions in gene expression regulation at the levels of both transcription and translation (Cohen et al., 2011; Guo and Shorter, 2017). Expressed in most human tissues and cell types, TDP-43 is predominantly non-phosphorylated and localized mostly within nuclei, while in disease states the protein is phosphorylated and often translocated to the cytoplasm (Neumann et al., 2006).
Following the detection of TDP-43 proteinopathy in FTLD-TDP and in the large majority of ALS cases (Mackenzie et al., 2007), TDP-43 proteinopathy was also discovered in the brains of subjects over age 80 years without FTLD or ALS, but often with comorbid hippocampal sclerosis and/or ADNC (Amador-Ortiz et al., 2007a, b). In subjects with ADNC, LATE-NC represents a common comorbid lesion that lowers the threshold for developing dementia (Josephs et al., 2014b, 2015). In retrospective studies, age-related TDP-43 proteinopathy has been associated with a progressive amnestic syndrome that mimicked the Alzheimer’s clinical syndrome (Pao et al., 2011; Brenowitz et al., 2014). TDP-43 proteinopathy, hippocampal sclerosis pathology, and the associated amnestic dementia increases with advanced age, while the prevalence of severe ADNC decreases in extreme old age (Nelson et al., 2011a, b, 2013; Brenowitz et al., 2014). The presence of pathological TDP-43 in these cases suggests a novel disease mechanism in older adults. As there is currently no universally agreed upon terminology or staging system for common age-related TDP-43 proteinopathy, this condition is under-studied and not well recognized even among investigators in the field of dementia research. The promotion of research and increasing awareness of this disease are the primary motivations for developing the new term LATE, and for the recommendations that follow.
LATE neuropathological changes
LATE-NC is a TDP-43 proteinopathy of advanced age, especially in subjects older than age 80. Following the convention proposed by a working group for the neuropathological criteria of Alzheimer’s disease (Montine et al., 2012), we use LATE to refer to the disease, and LATE-NC as the term to indicate LATE neuropathological changes. The term LATE is intended to encompass several previously used designations related to TDP-43 proteinopathy that may be associated with cognitive impairment, including hippocampal sclerosis, hippocampal sclerosis of ageing, hippocampal sclerosis dementia, cerebral age-related TDP-43 with sclerosis, and TDP-43 pathologies in the elderly (for reviews see Kuslansky et al., 2004; Lippa and Dickson, 2004; Nelson et al., 2013, 2016b; Dutra et al., 2015).
‘TDP-43 proteinopathy’ refers to loss of normal nuclear TDP-43 immunoreactivity, with TDP-43 protein ‘inclusion bodies’ in the neuronal cytoplasm, as well as abnormal TDP-43 accumulation (much of it phosphorylated) in nuclei and cell processes (neurites) of neurons and in oligodendroglia and astrocytes. For representative examples of TDP-43 proteinopathy, see Fig. 1. Mislocalized and phosphorylated TDP-43 is a necessary feature of LATE-NC and sometimes has characteristics similar to those seen in type A FTLD-TDP (Lin et al., 2009; Mackenzie et al., 2011; Murray et al., 2014; Aoki et al., 2015), but often the features do not fit cleanly into an established FTLD-TDP subtype. Indeed, a recent study described features of LATE-NC including both similarities and differences from type A FTLD-TDP (Josephs et al., 2019). In addition to limbic structures, TDP-43 proteinopathy in LATE has also been described in the olfactory bulb, neocortex, basal ganglia, and less frequently in brainstem (Josephs et al., 2008; Geser et al., 2010; Josephs and Dickson, 2016; Nelson et al., 2018). Immunoelectron microscopy showed that the TDP-43 inclusions have a fibrillary ultrastructure composed of bundled 10–20-nm diameter straight filaments (Lin and Dickson, 2008; Lin et al., 2009), often accompanied by electron dense granules (Cairns et al., 2007b; Robinson et al., 2013).
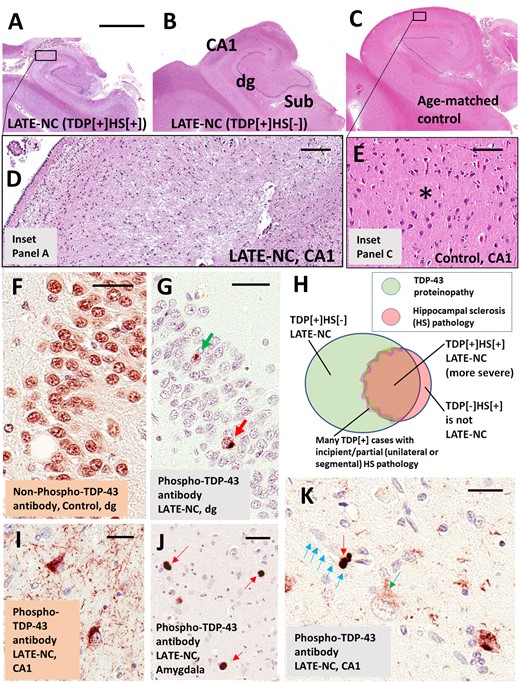
LATE neuropathological changes (LATE-NC). (A–E) Coronally sectioned human hippocampi stained using haematoxylin and eosin (H&E). Note that the photomicrographs in A–C are presented at the same magnification. (A) LATE-NC with hippocampal sclerosis (HS). The hippocampus is atrophic and the neuropil rarefied. (D) Higher magnification in CA1 subfield, with lack of normal cellular architecture and with extensive gliosis. (C) Control age-matched hippocampus. (E) CA1 of the control hippocampus to demonstrate the normal cellular architecture and intact eosinophilic neuropil (asterisk). The hippocampus shown in B is less atrophic, with less obvious neuropil disruption, in comparison to the case in A at low magnification; however, an adjacent section revealed TDP-43 proteinopathy. Hippocampal fields are labelled in B: dg = dentate granule layer; Sub = subiculum. TDP-43 proteinopathy can be recognized using antibodies raised against either non-phosphorylated or phosphorylated TDP-43 epitopes. (F) Dentate granule cells in a case lacking TDP-43 pathology. Note that cell nuclei are normally immunopositive for non-phosphorylated TDP-43 protein. In a case with LATE-NC (G), by contrast, an antibody against phosphorylated TDP-43 protein recognizes only the pathological inclusions in the nucleus (green arrow) and cytoplasm (red arrow). Unlike the antibody against non-phosphorylated TDP-43, the antibody against phosphorylated TDP-43 is negative in non-affected cells. Most cells in G are visualized with the counterstain, haematoxylin, which stains cell nuclei blue. The Venn diagram in H illustrates schematically the imperfect overlap between cases with TDP-43 proteinopathy, hippocampal sclerosis, and LATE-NC. A subset of cases with TDP-43 pathology have comorbid hippocampal sclerosis pathology; the change zone between non-hippocampal sclerosis and hippocampal sclerosis cases is indistinct because many cases seem to be in transition with incipient hippocampal neuron loss and gliosis. Importantly, cases with hippocampal sclerosis pathology but no TDP-43 proteinopathy (e.g. hippocampal sclerosis pathology associated with anoxia or epilepsy) are not classified as LATE-NC. (I) Phospho-TDP-43 proteinopathy in two neurons in hippocampal CA1, along with phospho-TDP-43 immunoreactive dystrophic neurites. (J) Tangle-like phospho-TDP-43 immunoreactive cytoplasmic inclusions in amygdala (red arrows) with fewer phospho-TDP-43 immunoreactive neurites in the background. (K) An intraneuronal phospho-TDP-43 inclusion (red arrow) and a phospho-TDP-43 deposit (green arrow) surrounding a capillary (shown with blue arrows); these TDP-43 immunoreactive structures have been demonstrated to exist within astrocyte end-feet (Lin et al., 2009). Note also the presence of a cell with cytoplasmic puncta (green arrow), perhaps in an early phase of phosphorylated TDP-43 proteinopathy. Scale bar in A = 4 mm for A–C; D = 200 μm; E = 100 μm; F = 30 μm; G = 35 μm; I = 30 μm; and K = 25 μm.
In brains with LATE-NC, haematoxylin and eosin stains may reveal neuronal dropout and astrocytosis in the CA1 sector of the hippocampus, as well as in the subiculum, entorhinal cortex, and amygdala (Amador-Ortiz and Dickson, 2008). Atrophy can be marked in these areas (Fig. 1A–C). In severely affected hippocampi, the neuropil becomes rarefied and loss of neuronal components is accompanied by reactive astrocytosis (Amador-Ortiz et al., 2007a). Pronounced leucocyte infiltrates or perivascular cuffing are not typically seen, but hypertrophic microglia can be numerous (Bachstetter et al., 2015). The neuronal cell loss is segmental in some subjects, observed in some but not all sections from the same brain area (Ighodaro et al., 2015). Hippocampal sclerosis pathology is unilateral in ∼40–50% of cases in which both sides were evaluated (Nelson et al., 2011b; Zarow et al., 2012; Kero et al., 2018), not unlike FTLD-TDP (Irwin et al., 2018).
Hippocampal sclerosis is present in a subset of cases with severe LATE-NC, and was the first characteristic pathological feature that distinguished it from ADNC (Dickson et al., 1994). Nevertheless, hippocampal sclerosis is neither specific to LATE-NC nor sufficient for the diagnosis of LATE. The neuropathological diagnosis of hippocampal sclerosis is fraught with difficulty. The most recent consensus guidelines for ADNC and related disorders stated that hippocampal sclerosis pathology is ‘defined by severe pyramidal cell loss and gliosis in CA1 and subiculum of the hippocampal formation that is out of proportion to AD neuropathologic change in the same structures’ (Montine et al., 2012). There is, however, significant topographic and phenotypic heterogeneity in hippocampal degeneration, creating difficulties in establishing strict criteria for widespread use. Moreover, hippocampal sclerosis is a pathological endpoint associated with various underlying disease processes, including epilepsy, hypoxia, hypoglycaemia, certain infections, and numerous neurodegenerative conditions (Josephs et al., 2007; Thom et al., 2009; Yokota et al., 2010; Malek-Ahmadi et al., 2013; Murray et al., 2013; Ling et al., 2017; Popkirov et al., 2017; Sen et al., 2018). Having originated in a 19th century study of epilepsy by Wilhelm Sommer (Sommer, 1880; Thom, 2009), the term hippocampal sclerosis is still used widely by radiologists and pathologists in the context of seizure disorders (Isnard and Bourdillon, 2015; Thom and Sisodiya, 2015). Detailed discussions of histopathological features and subtypes of hippocampal sclerosis can be found elsewhere (Probst et al., 2007; Rauramaa et al., 2013; Hatanpaa et al., 2014; Dutra et al., 2015; Thom and Sisodiya, 2015; Cykowski et al., 2017). Brains with hippocampal sclerosis, but lacking TDP-43 pathology (TDP-43−/HS+), do not represent LATE-NC. For example, brains with hippocampal sclerosis caused by acute hypoxia or associated with epilepsy are negative for TDP-43 proteinopathy (Amador-Ortiz et al., 2007b; Lee and Lee, 2008; Nelson et al., 2011b) and do not fulfil criteria for LATE-NC (Fig. 1H). In summary, TDP-43 proteinopathy is a necessary feature of LATE-NC that may or may not be accompanied by hippocampal sclerosis.
As has been the case in other neurodegenerative diseases (Braak et al., 1993, 2006; Thal et al., 2000; Zaccai et al., 2008; Alafuzoff et al., 2009), careful assessments of autopsy data, from both longitudinal studies of clinic-based research subjects as in the NIA-funded Alzheimer’s Disease Centers, and from community-based studies, have expanded our understanding of LATE. While subjects with advanced age and hippocampal sclerosis often have TDP-43 proteinopathy (Amador-Ortiz et al., 2007b; Nelson et al., 2011b; Robinson et al., 2014; Nag et al., 2015, 2018), TDP-43 proteinopathy in limbic structures is more prevalent than hippocampal sclerosis (Kovacs et al., 2013; Josephs et al., 2014b; Keage et al., 2014; Murray et al., 2014; Rahimi and Kovacs, 2014; Aoki et al., 2015; Nag et al., 2015, 2017; Hokkanen et al., 2018; Robinson et al., 2018b). The TDP-43-positive (+) and hippocampal sclerosis-negative (HS−) cases are a subset of LATE-NC that represent 5–40% of research subjects in autopsy series. Prior researchers have used terms for brains with TDP-43 proteinopathy and with some degree of cell dropout and gliosis, but lacking frank hippocampal sclerosis, as a ‘precursor to HS’, ‘pre-HpScl,’ or ‘pre-HS-Aging’ (Hatanpaa et al., 2008; Aoki et al., 2015; Hokkanen et al., 2018). As suggested by the terminology, TDP-43+/HS− brains may represent an early or transitional phase on the same disease continuum as TDP-43+/HS+ cases. There are other clues about LATE that were gathered from autopsy cohort studies. For example, even when hippocampal sclerosis was unilateral or segmental, the TDP-43 proteinopathy was almost always bilateral (Nelson et al., 2011b; Ighodaro et al., 2015). These observations have implications about how LATE evolves, which remains an important and open question.
Data gathered in large autopsy series have been analysed to test hypotheses about progression of LATE. Multivariable regression-based assessment can be used to generate models to test whether cross-sectional data align with proposed sequential pathways of neuropathological changes. Results of one such pathway analysis, from the Rush University community-based autopsy studies, are shown in Fig. 2. These analyses were performed as described previously (Power et al., 2018) and the findings are compatible with at least three hypotheses: (i) a subset of cases with TDP-43 proteinopathy develop hippocampal sclerosis caused or exacerbated by overlapping process(s) that promoted the TDP-43 proteinopathy, or directly by the TDP-43 proteinopathy itself; (ii) TDP-43 proteinopathy is associated independently with dementia, even in cases lacking hippocampal sclerosis; and (iii) pathogenetic mechanisms associated with ADNC (in Fig. 2, data are provided on neuritic amyloid-β plaques) are also associated with increased TDP-43 proteinopathy. Current rodent models of TDP-43 proteinopathy with hippocampal sclerosis-like pathology are few (Ke et al., 2015). TDP-43 proteinopathy was shown to be transmissible in mouse models similar to pathological tau and amyloid-β from Alzheimer’s disease brains (Porta et al., 2018), but the published TDP-43 models are thought to be more directly relevant to FTLD-TDP than LATE. For now, the lack of adequate longitudinal biomarker data and the limitations of current animal models hamper our study of disease mechanism(s) and further investigations are needed.
![Statistical analyses on data related to LATE from the Rush University community-based autopsy cohort depicting the results of pathway analyses. Data were analysed from research volunteers (total n = 1309) in two clinical‐pathological studies of ageing from Rush University as described previously (Power et al., 2018). In this sample, the mean age of death was 89.7 years [standard deviation (SD) 6.5 years, range 65–108 years]. These analyses incorporated age, density of amyloid-β neuritic amyloid plaques (to factor in ADNC), TDP-43 proteinopathy, hippocampal sclerosis pathology, and the endpoint of Alzheimer’s-type clinical dementia. The components of the pathway analyses most strongly associated with LATE-NC are shown in red. The numbers are path coefficients with standard error in parentheses (shown in purple). These numbers help to quantify the effects of individual pathways. For instance, the data are compatible with there being two pathways from TDP-43 proteinopathy to dementia, one direct pathway (TDP-43 proteinopathy→dementia) and the other indirect pathway that includes hippocampal sclerosis pathology (TDP-43 proteinopathy→hippocampal sclerosis→dementia): in the statistical model, the TDP-43 proteinopathy is independently associated with both hippocampal sclerosis pathology and clinical dementia status. Further, the data indicate that a subset of TDP-43 proteinopathy is ‘downstream’ of ADNC-type neuritic amyloid plaque pathology. In a practical sense, this means that brains with more neuritic amyloid plaques are more likely to have TDP-43 proteinopathy, with all other known factors being the same. Aβ = amyloid-β.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/142/6/10.1093_brain_awz099/1/m_awz099f2.jpeg?Expires=1716507675&Signature=D1dynLrM87aFspsoLoxKzkuwa6T951CTO-~r0YHq7pp8AXsf7oF4YG18412MJDgSp7CmxloGCZEl~HZKXFO4hpJ7CBb3hnBTkJ-7FTgwaO4T7rJQGZc3lIci7JZw10jYUtL6TxxGLLFVOWALjy4PUXZVY~Bo7jgxMyp-FyPL1gUpnp7dby6MOJkBOrilsQLFUjFJsKLH7FT9~4aNebK-pqyNSJmyJEtRZu8UPgUye2fZRbqS7VdyIr7Mk7yBif02aOVHfRa79rkG7ERW~IQPVZ0nHk-yF0MBILB-07Gi8jxftvvd-phhAzPUW0jpWgky5uI6actn67EqrrYtxxoEFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Statistical analyses on data related to LATE from the Rush University community-based autopsy cohort depicting the results of pathway analyses. Data were analysed from research volunteers (total n = 1309) in two clinical‐pathological studies of ageing from Rush University as described previously (Power et al., 2018). In this sample, the mean age of death was 89.7 years [standard deviation (SD) 6.5 years, range 65–108 years]. These analyses incorporated age, density of amyloid-β neuritic amyloid plaques (to factor in ADNC), TDP-43 proteinopathy, hippocampal sclerosis pathology, and the endpoint of Alzheimer’s-type clinical dementia. The components of the pathway analyses most strongly associated with LATE-NC are shown in red. The numbers are path coefficients with standard error in parentheses (shown in purple). These numbers help to quantify the effects of individual pathways. For instance, the data are compatible with there being two pathways from TDP-43 proteinopathy to dementia, one direct pathway (TDP-43 proteinopathy→dementia) and the other indirect pathway that includes hippocampal sclerosis pathology (TDP-43 proteinopathy→hippocampal sclerosis→dementia): in the statistical model, the TDP-43 proteinopathy is independently associated with both hippocampal sclerosis pathology and clinical dementia status. Further, the data indicate that a subset of TDP-43 proteinopathy is ‘downstream’ of ADNC-type neuritic amyloid plaque pathology. In a practical sense, this means that brains with more neuritic amyloid plaques are more likely to have TDP-43 proteinopathy, with all other known factors being the same. Aβ = amyloid-β.
LATE MRI studies
MRI studies have provided a complementary window into brain changes in LATE, highlighting brain atrophy both within and outside of the medial temporal lobes of brains with autopsy-verified LATE-NC. Prior studies featured research volunteers who underwent MRI with autopsy follow-up. Several of these studies focused on the subset of cases with hippocampal sclerosis (i.e. presumed severe LATE-NC), therefore, most of the published data were lacking information about less severely affected cases. With that caveat in mind, a common finding in MRI studies is that hippocampal atrophy is greater in cases with LATE-NC than in those with pure Alzheimer’s disease (Jagust et al., 2008; Josephs et al., 2008, 2017a; Dawe et al., 2011; Kaur et al., 2014; Dallaire-Theroux et al., 2017; Hanko et al., 2019). Barkhof et al. (2007) found that many subjects with medial temporal atrophy lacked primary underlying ADNC. In this study cohort, the sensitivity and specificity of severe atrophy for ADNC was 63% and 69%, respectively, consistent with prior findings (Jack et al., 2002). Josephs et al. (2008) reported that subjects with neuropathology consistent with LATE-NC tended to be older, with more cognitive impairment, and with more pronounced hippocampal atrophy than TDP-43− subjects. Zarow et al. (2011) also described atrophy and deformation of the hippocampus considerably greater in those with hippocampal sclerosis and LATE-NC than in those with only ADNC (Zarow et al., 2011). In hippocampal sclerosis associated with LATE-NC, hippocampal atrophy was often asymmetric, and it tended to progress in a rostral-caudal gradient in the hippocampus. Using post-mortem MRI, Dawe et al. (2011) reported stronger correlations between hippocampal atrophy and LATE-NC (with hippocampal sclerosis pathology) than between hippocampal atrophy and ADNC, and subjects with both ADNC and LATE-NC had greater hippocampal atrophy than those with only ADNC. A recent study found that the volume and shape of the amygdala is associated with underlying LATE-NC and that these structural changes are indicative of cognitive decline beyond what can be explained with other pathological indices (Makkinejad et al., 2019).
Post-mortem MRI research has also provided strong evidence that LATE-NC is associated with substantial brain atrophy outside the medial temporal lobes (Kotrotsou et al., 2015). Figure 3A shows updated data from the Rush University autopsy cohort. After controlling for demographics, ADNC and other age-related pathologies, LATE-NC was related to not only the mesial temporal lobe atrophy, but also to atrophy in the inferior frontal, anterior temporal, and insular cortices. It is noteworthy that this regional atrophy pattern corresponds with the distribution of TDP-43 proteinopathy at autopsy (Josephs et al., 2016; Nag et al., 2018) (Fig. 3B). These data are in agreement with pathological studies of LATE-NC, as well as neuroimaging in subjects with LATE-NC risk genotypes, showing widespread brain involvement (Neltner et al., 2014; Cykowski et al., 2016; Josephs et al., 2016; Nelson et al., 2016a; Nho et al., 2016).
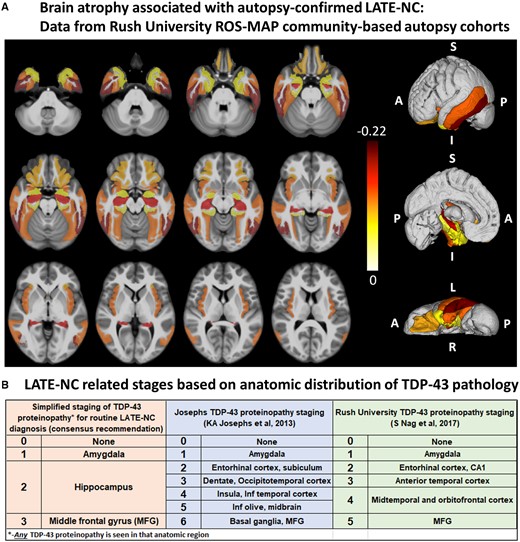
Brain regions that are affected in LATE. (A) Post-mortem MRI with autopsy confirmation allows discrimination of regions of brain atrophy associated with LATE-NC. These data indicate grey matter regions inside and outside of the medial temporal lobe with atrophy in cases with autopsy-confirmed LATE-NC from a community-based autopsy sample. The figure was prepared similarly to the methods used in Kotrotsou et al. (2015), with some modifications. Cerebral hemispheres from 539 participants of two cohort studies of ageing (Rush Memory and Aging Project and Religious Orders Study) were imaged with MRI ex vivo and also underwent detailed neuropathological characterization. The cortical and subcortical grey matter were segmented into 41 regions. Linear regression was used to investigate the association of regional volumes (normalized by height) with the score of LATE-NC at autopsy (scores: 0 = no TDP-43 inclusions, or inclusions in amygdala only; 1 = TDP-43 inclusions in amygdala as well as entorhinal cortex or hippocampus CA1, and neocortex; 2 = TDP-43 inclusions in amygdala, entorhinal cortex or hippocampus CA1, and neocortex, and hippocampal sclerosis pathology) controlling for amyloid plaques and neurofibrillary tangles, Lewy bodies, gross and microscopic infarcts, atherosclerosis, arteriolosclerosis, cerebral amyloid angiopathy, as well as age, sex, years of education, post-mortem interval to fixation and to imaging, and scanners. Unique colours have been assigned to different model estimates (units: mm2) for grey matter regions with significant negative correlation between their volumes and LATE pathology (P < 0.05, false discovery rate-corrected); darker colours indicate greater brain atrophy in that region. Results are overlaid on both hemispheres of the T1-weighted template of the IIT Human Brain Atlas (v.4.2). Lateral, medial and inferior to superior 3D views of the results are also shown. (B) Classification of LATE-NC according to anatomical region(s) affected by TDP-43 proteinopathy. The present working group recommended a simplified staging scheme for routine assessment of LATE-NC. This requires sampling and TDP-43 immunohistochemical staining of amygdala, hippocampus, and middle frontal gyrus. More detailed TDP-43 immunohistochemical staging schemes that are directly relevant to LATE-NC were previously published by Josephs et al. (2014a, 2016) and Nag et al. (2018). MFG = middle frontal gyrus.
Recommendations for routine autopsy evaluation and classification of LATE-NC
It is recommended that TDP-43 immunohistochemistry be performed as part of the neuropathological evaluation in all older subjects. At a minimum, immunohistochemical staining for TDP-43 is recommended in three brain areas: amygdala, mid-level hippocampus, and middle frontal gyrus. We recommend evaluating these regions as they are commonly obtained at autopsy of aged subjects and capture presumed progression of LATE-NC in the brain. This sampling includes the brain area affected early in the disease course (amygdala, Stage 1), an intermediate stage where the pathological change is robustly associated with cognitive impairment (hippocampus, Stage 2), and a region affected at more advanced stages (middle frontal gyrus, Stage 3) (Nag et al., 2018). Any detected TDP-43 proteinopathy is sufficient to define an anatomical region-based stage: for example, a minute amount of detected TDP-43 proteinopathy in the hippocampus indicates at least Stage 2. We emphasize that the proposed sampling for LATE-NC autopsy screening is a minimal evaluation, whereas more detailed sampling and staging should be considered for specific research settings (Josephs et al., 2014a, 2016; Uchino et al., 2015; Nag et al., 2017, 2018; Zhang et al., 2019). Figure 3B depicts staging schemes for LATE-NC, including sampling recommended for neuropathological evaluation of brain of older adults. This does not address regions that would be assessed in separate TDP-43 pathological staging schemes developed for ALS or FTLD-TDP (Brettschneider et al., 2013; Fatima et al., 2015; Tan et al., 2015; Verde et al., 2017; Neumann and Mackenzie, 2019).
Practical questions arise in relation to diagnostic ‘boundary zones’ between LATE-NC, FTLD-TDP, and ADNC. While both LATE-NC and FTLD-TDP may affect neocortical areas and may be comorbid with hippocampal sclerosis, LATE-NC usually has a later age of onset, an amnestic dementia, and limbic predominance of pathological change (Nelson et al., 2011b). On the other hand, recommendations for LATE-NC do not stipulate any age cut-offs, because the exact age ranges of disease susceptibility for FTLD-TDP or LATE-NC are not yet fully understood. For prior pathology-based comparisons between subtypes of TDP-43 proteinopathies (not related to age of onset), previous studies should be consulted (Amador-Ortiz et al., 2007a; Tan et al., 2015). More widespread and severe cortical atrophy is typically present in advanced FTLD-TDP than LATE-NC. There may indeed be features that could definitively distinguish LATE-NC cases (histopathologically or molecularly) from subtypes of FTLD-TDP (Arai et al., 2010; Hasegawa et al., 2011; Tsuji et al., 2012; Laferriere et al., 2019); however, more work is needed in this area. For now, definitive criteria to differentiate severe LATE-NC from FTLD-TDP await discovery of specific features that discriminate among various TDP-43 proteinopathies (Tan et al., 2017a).
Although LATE-NC and ADNC are recognized by differing neuropathological hallmarks, they may share upstream risk factors and disease mechanisms. Genetic variants predisposing to one protein misfolding disorder may also cause or exacerbate others (see below), and there may be interactions between the misfolded proteins themselves (Trojanowski and Lee, 2000; Higashi et al., 2007; Hu et al., 2008; Uryu et al., 2008; Kadokura et al., 2009; Davis et al., 2017; Spires-Jones et al., 2017; Tan et al., 2017b; Nelson et al., 2018). Brains that harbour ADNC, including some subjects with early-onset familial Alzheimer’s disease or Down syndrome, tend to also contain TDP-43 proteinopathy at rates higher than those lacking ADNC (Ala et al., 2000; Jellinger, 2000; Lippa et al., 2009; Davidson et al., 2011; Zarow et al., 2012). Individual neurons with both tau neurofibrillary tangle pathology and TDP-43 inclusions have been described, particularly in the amygdala, entorhinal cortex, and dentate gyrus of the hippocampus (Amador-Ortiz et al., 2007b; Kadokura et al., 2009; Smith et al., 2017; Robinson et al., 2018c; Josephs et al., 2019). Several published accounts have evaluated the connections between primary age-related tauopathy (PART) and age-related TDP-43 proteinopathy (Josephs et al., 2017b; Smith et al., 2017; Zhang et al., 2019), and TDP-43 proteinopathy has also been described in brains with coexisting argyrophilic grains or glial tauopathy (Fujishiro et al., 2009; Yokota et al., 2010; Arnold et al., 2013; Kertesz et al., 2015). The implications of comorbid amyloid-β and various tau pathologies in the context of LATE-NC are still incompletely understood, so further studies are required. There is also evidence that Lewy body disease and TDP-43 proteinopathy may coexist (Nakashima-Yasuda et al., 2007; McAleese et al., 2017; Miki et al., 2018; Trieu et al., 2018). On the other hand, many cases with ‘end-stage’ ADNC or Lewy body disease lack TDP-43 proteinopathy, so we recommend reporting the presence or absence of LATE-NC as a separate diagnostic entity, even when there are comorbid amyloid-β, tau and/or α-synuclein proteinopathies.
Additional research is required to guide future consensus-based recommendations in this evolving field. In terms of immunohistochemical reagents used to detect TDP-43 proteinopathy, there is no current consensus that a specific antibody can be recommended. Many neuropathologists use sensitive phospho-TDP-43 antibodies (Hasegawa et al., 2008; Alafuzoff et al., 2015); small aggregates can be readily seen using these reagents. Others use antibodies against non-phosphorylated epitopes, especially for detecting early changes (pathological nuclear to cytoplasmic redistribution) that may precede inclusion body formation (Vatsavayai et al., 2016; Braak et al., 2017; Braak and Del Tredici, 2018; Nana et al., 2019). It is unclear whether the absence of nuclear TDP-43 is reversible, but animal studies using inducible pathogenetic systems would suggest so (Ke et al., 2015). Further, there is some evidence that TDP-43 antigenicity can be vulnerable to fixation artefacts, and epitope retrieval methodology can influence results (Hatanpaa et al., 2008). Additional practice guidelines for studying LATE-NC need formal blinded cross validation studies as has been done for amyloid-β, tau and α-synuclein pathological biomarkers. Future studies will be needed to validate and refine systems for staging LATE-NC, and grading local pathological severity, as they relate to clinical and neuroimaging outcomes, especially since at least three staging schemes have been proposed as summarized in Fig. 3B.
Clinical and neurocognitive features of LATE
The clinical course of subjects with autopsy-proven LATE-NC has been characterized as an amnestic cognitive syndrome that can evolve to incorporate multiple cognitive domains and ultimately to impair activities of daily living, i.e. the dementia syndrome (Nelson et al., 2010; Nag et al., 2015; Robinson et al., 2018a, b). The cognitive impairment is greater than can be accounted for by ADNC or other pathologies (Gold et al., 2000; Kawas and Corrada, 2006; Imhof et al., 2007; Giannakopoulos et al., 2008; Nelson et al., 2011b; Kravitz et al., 2012; Boyle et al., 2013; Erten-Lyons et al., 2013). Initial reports on subjects with LATE-NC were focused on subjects with severe pathology (Dickson et al., 1994; Snowdon et al., 1997; Crystal et al., 2000; Vinters et al., 2000; Leverenz et al., 2002; Kuslansky et al., 2004; Zarow et al., 2005, 2008; Attems and Jellinger, 2006; Chui et al., 2006; Leverenz and Lipton, 2008), which helped to show that LATE-NC can be associated with dementia. More recent autopsy series, with both large sample sizes and broad ranges of clinical and pathological findings, have enabled statistical approaches to model the likely relative impact of each disease type. With these methods, LATE-NC was associated with substantial cognitive impairment that was independent of other coexisting pathologies (Nelson et al., 2010; Keage et al., 2014; Murray et al., 2014; Josephs et al., 2015; Nag et al., 2017). Table 1 shows primary data on the relationship between LATE-NC (stratified by the recommended three-stage system) and cognition. The neurological features associated with LATE-NC were different from the behavioural or aphasic clinical syndromes seen in FTLD-TDP cases (Nelson et al., 2011b; Jung et al., 2014; Wilson et al., 2019). While TDP-43 proteinopathy has been documented in some cognitively unimpaired subjects (Arnold et al., 2013; Keage et al., 2014; Uchino et al., 2015; Elobeid et al., 2016; Nascimento et al., 2016; Nag et al., 2018; Nascimento et al., 2018), it is likely that this represents preclinical disease in subjects dying before onset of clinical symptoms; such clinical resilience to pathological changes has been described in many disorders (Perkins et al., 2003; Shojania et al., 2003; Roulson et al., 2005; Latimer et al., 2017; Robinson et al., 2018b).
Selected parameters from a large community-based autopsy cohort, stratified by LATE-NC stages
| Characteristics | Consensus proposed LATE-NC TDP-43 stages | P-value | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| n | 666 | 263 | 258 | 189 | - |
| Age at death (SD) | 87.9 (6.8) | 89.9 (6.2) | 91.8 (5.6) | 91.9 (5.4) | <0.001 |
| % Female | 65.3 | 67.7 | 74 | 72.5 | 0.040 |
| Clinical diagnosis | <0.001 | ||||
| % Normal | 41.8 | 33.5 | 18.9 | 7.6 | |
| % MCI or dementia | 58.2 | 66.5 | 81.1 | 92.4 | |
| % with comorbid HS pathology | 1.7 | 3.5 | 13.6 | 42.9 | <0.001 |
| Cognitive function tests proximate to death, mean (SD) | |||||
| MMSE score | 22.8 (8.1) | 21.2 (8.9) | 18.2 (9.8) | 14.0 (10.0) | <0.001 |
| Episodic memory score | −0.60 (1.28) | −0.76 (1.31) | −1.36 (1.34) | −2.06 (1.23) | <0.001 |
| Characteristics | Consensus proposed LATE-NC TDP-43 stages | P-value | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| n | 666 | 263 | 258 | 189 | - |
| Age at death (SD) | 87.9 (6.8) | 89.9 (6.2) | 91.8 (5.6) | 91.9 (5.4) | <0.001 |
| % Female | 65.3 | 67.7 | 74 | 72.5 | 0.040 |
| Clinical diagnosis | <0.001 | ||||
| % Normal | 41.8 | 33.5 | 18.9 | 7.6 | |
| % MCI or dementia | 58.2 | 66.5 | 81.1 | 92.4 | |
| % with comorbid HS pathology | 1.7 | 3.5 | 13.6 | 42.9 | <0.001 |
| Cognitive function tests proximate to death, mean (SD) | |||||
| MMSE score | 22.8 (8.1) | 21.2 (8.9) | 18.2 (9.8) | 14.0 (10.0) | <0.001 |
| Episodic memory score | −0.60 (1.28) | −0.76 (1.31) | −1.36 (1.34) | −2.06 (1.23) | <0.001 |
Data were analysed from the Rush University ROS-MAP community-based autopsy cohort; n = 1376.
These data were analysed as described previously (Nag et al., 2018) from the Rush University Religious Orders Study (ROS), showing clinical, pathological, and cognitive status features. The new consensus guidelines for LATE-NC staging were applied to highlight the associations between LATE-NC severity (operationalized with new recommended staging method) including hippocampal sclerosis (HS) pathology and cognitive function tests. Note that many of these subjects in all the LATE-NC stages have additional pathologies including ADNC as described previously (Schneider et al., 2007; James et al., 2016).
MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination.
Selected parameters from a large community-based autopsy cohort, stratified by LATE-NC stages
| Characteristics | Consensus proposed LATE-NC TDP-43 stages | P-value | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| n | 666 | 263 | 258 | 189 | - |
| Age at death (SD) | 87.9 (6.8) | 89.9 (6.2) | 91.8 (5.6) | 91.9 (5.4) | <0.001 |
| % Female | 65.3 | 67.7 | 74 | 72.5 | 0.040 |
| Clinical diagnosis | <0.001 | ||||
| % Normal | 41.8 | 33.5 | 18.9 | 7.6 | |
| % MCI or dementia | 58.2 | 66.5 | 81.1 | 92.4 | |
| % with comorbid HS pathology | 1.7 | 3.5 | 13.6 | 42.9 | <0.001 |
| Cognitive function tests proximate to death, mean (SD) | |||||
| MMSE score | 22.8 (8.1) | 21.2 (8.9) | 18.2 (9.8) | 14.0 (10.0) | <0.001 |
| Episodic memory score | −0.60 (1.28) | −0.76 (1.31) | −1.36 (1.34) | −2.06 (1.23) | <0.001 |
| Characteristics | Consensus proposed LATE-NC TDP-43 stages | P-value | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| n | 666 | 263 | 258 | 189 | - |
| Age at death (SD) | 87.9 (6.8) | 89.9 (6.2) | 91.8 (5.6) | 91.9 (5.4) | <0.001 |
| % Female | 65.3 | 67.7 | 74 | 72.5 | 0.040 |
| Clinical diagnosis | <0.001 | ||||
| % Normal | 41.8 | 33.5 | 18.9 | 7.6 | |
| % MCI or dementia | 58.2 | 66.5 | 81.1 | 92.4 | |
| % with comorbid HS pathology | 1.7 | 3.5 | 13.6 | 42.9 | <0.001 |
| Cognitive function tests proximate to death, mean (SD) | |||||
| MMSE score | 22.8 (8.1) | 21.2 (8.9) | 18.2 (9.8) | 14.0 (10.0) | <0.001 |
| Episodic memory score | −0.60 (1.28) | −0.76 (1.31) | −1.36 (1.34) | −2.06 (1.23) | <0.001 |
Data were analysed from the Rush University ROS-MAP community-based autopsy cohort; n = 1376.
These data were analysed as described previously (Nag et al., 2018) from the Rush University Religious Orders Study (ROS), showing clinical, pathological, and cognitive status features. The new consensus guidelines for LATE-NC staging were applied to highlight the associations between LATE-NC severity (operationalized with new recommended staging method) including hippocampal sclerosis (HS) pathology and cognitive function tests. Note that many of these subjects in all the LATE-NC stages have additional pathologies including ADNC as described previously (Schneider et al., 2007; James et al., 2016).
MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination.
Although there is overlap in clinical features of autopsy-confirmed LATE-NC and ADNC (Pao et al., 2011; Brenowitz et al., 2014; Murray et al., 2014; Nag et al., 2017), careful analyses may identify distinctive neurocognitive features. Preliminary evidence suggests that subjects with relatively ‘pure’ LATE-NC (lacking severe comorbid pathologies) tend to have a more gradual clinical decline compared to those with ‘pure’ ADNC (Murray et al., 2014; Boyle et al., 2017). In contrast, those with comorbid ADNC and LATE-NC showed faster decline and more severe cognitive impairment than those with either ADNC or LATE-NC alone (Josephs et al., 2014b, 2015; Nag et al., 2017). In studies with both detailed longitudinal cognitive testing and comprehensive neuropathological evaluations, subjects with LATE-NC had prominent impairment in episodic memory (Table 1), but other cognitive domains and global cognitive status were also commonly affected especially in the later disease stages (Nag et al., 2015, 2017, 2018; Wilson et al., 2019). Correlative studies indicate that certain neurocognitive assessments, such as verbal fluency measures, are not independently associated with hippocampal volume, but are instead correlated with neocortical grey matter volumes (Ajilore et al., 2015; Pelletier et al., 2017). Correspondingly, subjects with relatively preserved verbal fluency (cortically-dependent), despite profound deficiency in word list delayed recall (hippocampal-dependent), have been shown to be at risk for LATE-NC (Nelson et al., 2011b). This pattern of neurocognitive test scores in LATE differs from that seen in subjects with ADNC alone (Nelson et al., 2011b) or FTLD-TDP (Brenowitz et al., 2014).
Neuropsychiatric disturbances have been reported in some subjects with LATE-NC (Ighodaro et al., 2015), and a retrospective, cross-sectional, multicentre study found evidence of increased risk of ‘agitation/aggression’ symptoms in subjects with ADNC and comorbid TDP-43 proteinopathy in comparison to subjects with ADNC lacking TDP-43 proteinopathy (Sennik et al., 2017). However, not all prior studies found that LATE-NC was associated with non-amnestic manifestations (Velakoulis et al., 2009; Nelson et al., 2011b; Vatsavayi et al., 2014; Sahoo et al., 2018). Future investigations are warranted to test for specific neuropsychiatric, motor, or autonomic signs that distinguish LATE from other degenerative disorders.
Public health impact of LATE
The public health impact of LATE is likely to be quite significant. Two basic study design elements that influence recognition of LATE-NC in autopsy cohorts are the age range in the cohort, and the date of the study. Researchers were unaware of TDP-43 proteinopathy prior to 2006, so studies prior to this time could not assess the specific impact of LATE. LATE-NC is mostly seen in the oldest-old, whereas in early clinical-pathological correlation studies of dementia (Roth et al., 1966; Blessed et al., 1968), the research subjects had died in their early 70s. LATE-NC needs to be assessed in population studies that include all age ranges. More recent clinical studies have demonstrated biomarker evidence of ‘suspected non-Alzheimer’s disease pathophysiology’ (SNAP) causing amnestic type cognitive impairment with substantial hippocampal atrophy but lacking detectable amyloid-β amyloidosis (Caroli et al., 2015; Burnham et al., 2016; Jack et al., 2016, 2017; Abner et al., 2017; Wisse et al., 2018). For example, the evaluation of 1535 participants in the Mayo Clinic Study of Aging showed significantly greater prevalence of SNAP compared with preclinical Alzheimer’s disease, and multimorbidity was increased in SNAP (odds ratio 2.16) (Vassilaki et al., 2018). LATE is probably an important contributor in this group of subjects (see below).
Among subjects autopsied past 80 years of age, most studies indicate that >20% of brains had pathological features consistent with LATE-NC (Fig. 4). It is noteworthy that the majority of these cases had additional comorbid pathologies, so the measured clinical-pathological correlation (relative contribution of each pathology to cognitive impairment) depends on how the investigators defined diagnostic thresholds and cut-points. The frequency of LATE-NC in autopsy series have varied, ranging from 5% to 50% of brains that were evaluated using TDP-43 immunohistochemistry, approximately twice the frequencies that were detected in prior studies that could only assess hippocampal sclerosis pathology (Leverenz et al., 2002; Lippa and Dickson, 2004; Arai et al., 2009; Nelson et al., 2011b; Rauramaa et al., 2011; Tremblay et al., 2011; Corrada et al., 2012; Zarow et al., 2012; Malek-Ahmadi et al., 2013; Keage et al., 2014; Jellinger and Attems, 2015; Uchino et al., 2015; Takao et al., 2016; Latimer et al., 2017; McAleese et al., 2017; Hokkanen et al., 2018; Kero et al., 2018; Robinson et al., 2018a). Differences in study design, including the application of various criteria for defining pathological abnormalities, pathological methods, recruitment strategy, and cohort demographics, all contribute to the variability in the reported frequency of LATE-NC.
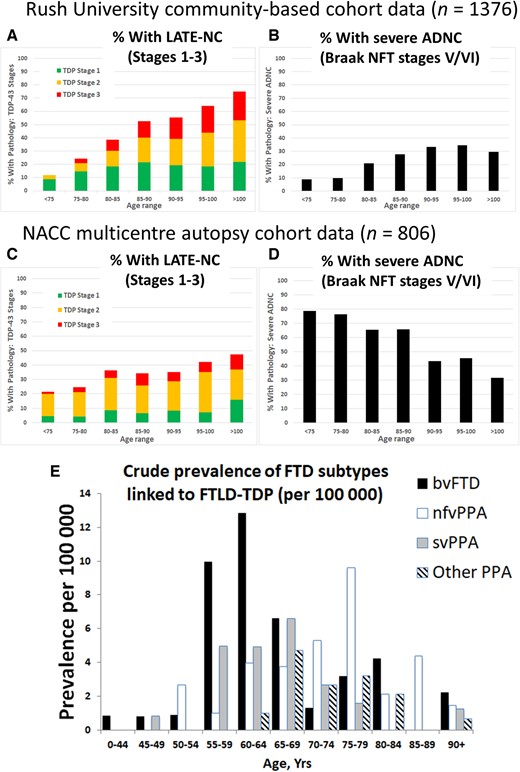
Different neurodegenerative disease conditions stratified by age: LATE-NC, severe ADNC, and FTD. FTD/FTLD cases were not present in data shown in A–D. Note that published studies to estimate disease prevalence for the various diseases have used importantly different study designs—thus, E is a clinical (no autopsy) study because population-based autopsy cohorts lack substantial numbers of FTD/FTLD cases. (A and B) Data from a community-based autopsy cohort—the Rush University ROS-MAP cohort (overall n = 1376). The TDP-43 pathology is operationalized using standard methods as described previously (Nag et al., 2018) and then the current paper’s suggested simplified staging system was applied; sample sizes for each age group (in years) are: <75 (n = 34); 75–80 (n = 82); 80–85 (n = 192); 85–90 (n = 375); 90–95 (n = 407); 95–100 (n = 222); and >100 (n = 64). Note that in this community-based sample, the proportion of cases with advanced ADNC is <50% in all age groups. (C and D) Data from the National Alzheimer’s Coordinating Center (NACC), which derives from 27 different research centres, as described previously (Besser et al., 2018; Katsumata et al., 2018). Overall sample size is n = 806, stratified thus by age groups (in years): <75 (n = 155); 75–80 (n = 118); 80–85 (n = 165); 85–90 (n = 170); 90–95 (n = 122); 95–100 (n = 57); and >100 (n = 19). The NACC research subjects were more likely to come to autopsy after being followed in dementia clinics, and the sample includes a higher percentage of subjects with severe ADNC. The percentage of subjects with LATE-NC is still >20% in each age group. Note that in both the community-based cohort (A and B) and clinic-based cohort (C and D), the proportion of subjects with severe ADNC decreased in advanced old age, while in the same cases the proportion of subjects with LATE-NC increased. (E) Epidemiological data on FTD syndromes for comparison to LATE. Data are provided about crude prevalence rates for FTD syndromes that have been associated with FTLD-TDP. Several of these clinical syndromes are likely to have considerable numbers of cases with FTLD-tau (bvFTD and nfvPPA) or ADNC (other PPA) rather than FTLD-TDP, so the actual prevalence of FTLD-TDP pathology is probably lower than these data suggest. Note that the clinical syndromes associated with FTLD-TDP have a prevalence that are several orders of magnitude lower than LATE-NC. These data, described in detail previously (Coyle-Gilchrist et al., 2016), derive from multisource referral over 2 years, which identified all diagnosed or suspected cases of FTD subtypes in two UK counties comprising the PiPPIN (Pick’s Disease and Progressive Supranuclear Palsy: Prevalence and Incidence) catchment area in the East of England. Two cities in the PiPPIN catchment area were Norfolk and Cambridge. Diagnostic confirmation used current consensus diagnostic criteria after interview and re-examination. Total sample size was n = 986 483 subjects. Shown are crude prevalence rates for the major FTLD-TDP associated syndromes by age and syndrome. bvFTD = behavioural variant frontotemporal dementia; nfvPPA = non-fluent agrammatic variant primary progressive aphasia; svPPA = semantic variant PPA. Note that subjects between ages 55 and 80 are at greatest risk for FTD, and, the FTLD-TDP associated FTD syndrome prevalence is <30 per 100 000 (E), in sharp contrast to the data shown in A–D.
One approach that can be used to assess the public health impact of a disease is the evaluation of attributable risk (Porta, 2014). Although generally used to study the impact of risk factors on disease prevalence in a population (Bruzzi et al., 1985), this statistical method can be applied to neuropathological studies to indicate the relative impact of different neuropathologies on clinical dementia. More specifically, the assessment of attributable risk can query how the frequency of LATE-NC, in relation to other common brain lesions detected at autopsy, is associated with the probability of a dementia diagnosis (Boyle et al., 2019). This analytical approach theoretically makes it possible to estimate the proportion of dementia that might be prevented if LATE-NC could be eliminated, and to compare that with other neuropathologies. The results of an analysis of attributable risk in the Rush University Religious Orders Study is shown in Table 2; methods have been described in detail previously (Boyle et al., 2019). These data are compatible with the hypothesis that a significant (∼15–20%) proportion of clinically diagnosed Alzheimer’s disease dementia (i.e. the Alzheimer’s clinical syndrome) in advanced age is attributable to LATE-NC; the impact is about half the magnitude of ADNC in this group of older subjects, and the impact is similar to the combined effects of vascular neuropathologies.
A statistical analysis of attributable risk from research volunteers in two clinical‐pathological studies of ageing from Rush University
| Neuropathological indices | Fraction attributable % (95% CI)a |
|---|---|
| Alzheimer’s disease (ADNC) | 39.4 (31.5–47.4) |
| Vascular disease pathologyb | 24.8 (17.3–32.1) |
| LATE-NC | 17.3 (13.1–22.0) |
| α-Synucleinopathy/Lewy body pathology | 11.9 (8.4–15.6) |
| Neuropathological indices | Fraction attributable % (95% CI)a |
|---|---|
| Alzheimer’s disease (ADNC) | 39.4 (31.5–47.4) |
| Vascular disease pathologyb | 24.8 (17.3–32.1) |
| LATE-NC | 17.3 (13.1–22.0) |
| α-Synucleinopathy/Lewy body pathology | 11.9 (8.4–15.6) |
Shown are fractions of dementia of the Alzheimer type cases that were attributable to individual neuropathological indices in advanced age. In this sample, the mean age of death was 89.7 years (SD 6.5 years, range 65–108 years). For these analyses, multivariable logistic regression models examined associations of neuropathological indices with the outcome of Alzheimer’s-type clinical dementia and quantified the percentage of cases attributable to each. Methods have been described in detail previously (Boyle et al., 2019). These data give strong indication that the public health impact of LATE is large, on the same order of magnitude as ADNC, vascular pathologies, and Lewy body pathology.
a95% CIs were derived using bootstrapping.
bVascular pathologies included: cerebral amyloid angiopathy, atherosclerosis, arteriolosclerosis and gross infarcts.
A statistical analysis of attributable risk from research volunteers in two clinical‐pathological studies of ageing from Rush University
| Neuropathological indices | Fraction attributable % (95% CI)a |
|---|---|
| Alzheimer’s disease (ADNC) | 39.4 (31.5–47.4) |
| Vascular disease pathologyb | 24.8 (17.3–32.1) |
| LATE-NC | 17.3 (13.1–22.0) |
| α-Synucleinopathy/Lewy body pathology | 11.9 (8.4–15.6) |
| Neuropathological indices | Fraction attributable % (95% CI)a |
|---|---|
| Alzheimer’s disease (ADNC) | 39.4 (31.5–47.4) |
| Vascular disease pathologyb | 24.8 (17.3–32.1) |
| LATE-NC | 17.3 (13.1–22.0) |
| α-Synucleinopathy/Lewy body pathology | 11.9 (8.4–15.6) |
Shown are fractions of dementia of the Alzheimer type cases that were attributable to individual neuropathological indices in advanced age. In this sample, the mean age of death was 89.7 years (SD 6.5 years, range 65–108 years). For these analyses, multivariable logistic regression models examined associations of neuropathological indices with the outcome of Alzheimer’s-type clinical dementia and quantified the percentage of cases attributable to each. Methods have been described in detail previously (Boyle et al., 2019). These data give strong indication that the public health impact of LATE is large, on the same order of magnitude as ADNC, vascular pathologies, and Lewy body pathology.
a95% CIs were derived using bootstrapping.
bVascular pathologies included: cerebral amyloid angiopathy, atherosclerosis, arteriolosclerosis and gross infarcts.
Also pertinent to the current and future public health impact of LATE is the age range of subjects with highest risk for the disease. The tendency for LATE-NC to occur among the oldest-old has been appreciated for decades, since the groundbreaking studies on age-related hippocampal sclerosis (Crystal et al., 1993; Dickson et al., 1994), a pathological manifestation later shown to be associated with LATE-NC. In multiple subsequent large autopsy samples, LATE-NC was observed with increasing frequency in each year of life after age 85 (Nelson et al., 2013; Keage et al., 2014; Uchino et al., 2015; Hokkanen et al., 2018) (Fig. 4). This is in contrast to amyloid-β plaques, which are common (seen in >50% of subjects) in all elderly age groups, but are not universal and not more frequently seen at autopsy with every year of advanced old age (Braak et al., 2011; Nelson et al., 2011a, 2013; Brenowitz et al., 2014; Neltner et al., 2016). LATE appears to be ∼100-fold more prevalent than FTD syndromes, which tend to affect younger subjects (Knopman and Roberts, 2011; Coyle-Gilchrist et al., 2016) (Fig. 4E; note the y-axis scale). Females are generally more likely to survive to advanced old age than males (Neltner et al., 2016), which places them at increased lifetime risk for LATE. Otherwise, there is no compelling evidence to date of strong sex-related or ethnoracial differences in susceptibility to LATE (Brenowitz et al., 2014; Murray et al., 2014; Latimer et al., 2017; Oveisgharan et al., 2018), but further studies in diverse populations are needed. Since most relevant current data were derived from autopsy cohorts, the prevalence of LATE-NC may be higher in younger subjects than currently recognized if there is a survival bias (those with LATE-NC live to older ages), underscoring the need for more longitudinal studies that incorporate clinical biomarkers. Since the demographic group made up of subjects past 85 years of age is predicted to greatly expand in the coming decades (Gardner et al., 2013; Nelson et al., 2013), LATE is likely to become a far greater public health burden in the future unless preventative or therapeutic strategies are developed.
Genetics of LATE
Genetic studies provide insights into disease-related mechanisms and, potentially, future therapeutic targets. The following five genes (in the chronological order in which they were identified) have been reported to harbour risk alleles associated with pathological manifestations we refer to as LATE-NC: granulin (GRN) on chromosome 17q, transmembrane protein 106B (TMEM106B) on chromosome 7p, ATP-binding cassette sub-family member 9 (ABCC9) on chromosome 12p, potassium channel subfamily M regulatory beta subunit 2 (KCNMB2) on chromosome 3q, and apolipoprotein E (APOE) on chromosome 19q (Dickson et al., 2010; Pao et al., 2011; Beecham et al., 2014; Murray et al., 2014; Nelson et al., 2014, 2015b; Aoki et al., 2015; Katsumata et al., 2017; Yang et al., 2018). See Supplementary Table 1 for summary information on these genes and their associated phenotypes. For this discussion, we include the endophenotype that was used in the published research (usually hippocampal sclerosis) rather than LATE-NC.
Gene variants in GRN and TMEM106B were shown to be associated with hippocampal sclerosis and TDP-43 proteinopathy risk using allele tests, based on the known relationship of those two genes to FTLD-TDP (Baker et al., 2006; Boeve et al., 2006; Cruts et al., 2006; Van Deerlin et al., 2010). These gene variants have now been most consistently associated with risk of LATE-NC. For the association between the GRN and hippocampal sclerosis, Dickson et al. showed that hippocampal sclerosis in aged subjects was associated with the T-allele of the GRN single nucleotide polymorphism (SNP) rs5848 (Dickson et al., 2010; Murray et al., 2014). Aoki and colleagues reported that the frequency of the C-allele of TMEM106B rs1990622 in hippocampal sclerosis was lower than that in non-hippocampal sclerosis controls (Aoki et al., 2015). Following the initial studies, the findings were replicated of increased risk for hippocampal sclerosis associated with each copy of the T-allele of TMEM106B rs1990622 (Nelson et al., 2014, 2015b; Dickson et al., 2015; Yu et al., 2015).
Since GRN and TMEM106B were both implicated in FTLD-TDP, their strong association with LATE-NC provides compelling evidence for pathogenetic overlap between FTLD-TDP and LATE. From a mechanistic perspective, the cognate proteins for these genes have been shown to play important roles in endosomal/lysosomal biology, and there is experimental evidence for interaction of these gene products (Chen-Plotkin et al., 2012; Nicholson and Rademakers, 2016; Klein et al., 2017; Zhou et al., 2017; Paushter et al., 2018). The TMEM106B gene appears to be pleiotropic for multiple diseases (Gallagher et al., 2014; Ou et al., 2015; Hsiao et al., 2017; Cherry et al., 2018; Chornenkyy et al., 2019), and the LATE-NC risk allele in TMEM106B may influence healthy brain ageing (Rhinn and Abeliovich, 2017; Ren et al., 2018). Separate studies have found that GRN gene products (granulins) play roles in inflammation and wound repair (Ahmed et al., 2007; Miller et al., 2013). Notably, the GRN risk variant rs5848 has been associated with increased inflammatory mediators in CSF (e.g. AXL and CLU) (Fardo et al., 2017). More work is required to enable better understanding of how molecular pathways relevant to FTLD-TDP are involved in LATE.
An important recent finding by several different groups is that the APOE ɛ4 allele, which is a risk factor for ADNC and Lewy body disease, is also associated with increased risk for TDP-43 proteinopathy in the elderly (Robinson et al., 2018c; Wennberg et al., 2018; Yang et al., 2018). Other studies did not find an association between APOE genotypes and risk for hippocampal sclerosis (Troncoso et al., 1996; Leverenz et al., 2002; Nelson et al., 2011b; Pao et al., 2011; Brenowitz et al., 2014; Hall et al., 2019; but see Farfel et al., 2016). Few subjects with the APOE ɛ4 allele survive into advanced old age without any amyloid-β plaques (Saunders et al., 1993; Schmechel et al., 1993), and it remains to be seen exactly how the APOE ɛ4 protein influences TDP-43 proteinopathy. Nevertheless, recent studies from large research cohorts have provided additional insights into the presence of pathogenetic mechanisms that are shared between neurodegenerative diseases.
Since the presence or absence of risk alleles in TMEM106B, GRN, and APOE cannot by themselves or in combination confidently predict the risk for LATE-NC in a given subjects (Katsumata et al., 2017; Nelson et al., 2019), there must be other factors that influence the disease phenotype. The connections of the ABCC9 and KCNMB2 genes with risk of LATE-NC were discovered via genome-wide association studies (GWAS), which are neither helped nor biased by prior mechanistic hypotheses. The finding of the associations between ABCC9 gene variants and LATE-NC (Nelson et al., 2015b), and brain atrophy detected with MRI (Nho et al., 2016), were reported in separate samples from the initial GWAS (Nelson et al., 2014). Neither ABCC9 nor KCNMB2 gene variants were associated with LATE-NC in cohorts other than those described above. ABCC9 and KCNMB2 are genes coding for proteins that serve to regulate potassium channels (Zarei et al., 2007; Nelson et al., 2015a). The ABCC9 risk genotype also implicates thyroid hormone dysregulation in LATE-NC; the locus was found to be associated with altered brain expression of genes induced by thyroid hormone (Nelson et al., 2016a). Thyroid hormones have been found to be dysregulated in subjects with autopsy-confirmed LATE-NC in recent studies (Trieu et al., 2018; Nelson et al., 2019), and high thyrotropin was associated with reduced hippocampal volume in a population-based study (Ittermann et al., 2018). A gene variant near ABCC9, which lies within both the SLCO1A2 and IAPP genes, was also found in a GWAS study to be associated with neurodegeneration disproportional to amyloid-β accumulation (Roostaei et al., 2016), which may indicate LATE in those cases. The KCNMB2 gene has been associated with suicidal ideation in US military veterans (Kimbrel et al., 2018) and may be related to depression, which is common in the elderly. Further, when KCNMB2 is overexpressed in the hippocampus of mice, it rescues memory deficits (Yu et al., 2018). More work is required to enable better understanding and identification of the molecular pathways involved in LATE.
Prior genetic studies on TDP-43 proteinopathy and hippocampal sclerosis have varied in important ways, including patient inclusion/exclusion criteria, disease definitions, and age composition, which may explain their differing findings with regard to genotype/phenotype associations. The prospects for successful future genetic discoveries will be improved by the development of specific and standardized LATE-NC endophenotypes. FTLD-TDP provides an example in which pathological subtyping of patients has been beneficial for genetic correlation studies: there are, for example, strong associations between TMEM106B and GRN gene variants with FTLD-TDP type A pathology (Rademakers et al., 2008; Aoki et al., 2015). Preliminary studies suggest that distinguishing morphology of TDP-43 pathology in LATE-NC may also be relevant to genetic risk (Josephs et al., 2019). We speculate that genetic profiling may eventually become a key consideration for recruitment to clinical trials, and possible future precision medicine approaches, since some genotypes may be differentially responsive to specific interventions.
LATE biomarkers
Optimal biomarkers for LATE, including biofluids or PET ligands, would be specific for the disease-defining feature, namely TDP-43 proteinopathy (Steinacker et al., 2018). At this time, no biofluid or PET biomarker satisfies this essential criterion of molecular specificity. Nor do PET ligands for LATE seem to be on the near-term horizon. The problems of intracellular location and small pathological burden of TDP-43 proteinopathy are obstacles that limit signal-to-noise ratio for biomarkers.
The NIA-AA Research Framework group recommended a system for classifying subjects based on amyloid-β amyloid (A), tau (T) and neurodegeneration/neuronal injury (N) biomarkers, which is termed AT(N) (Burnham et al., 2016; Jack et al., 2016). Each biomarker category can be binarized as positive (+) or negative (−) resulting in eight possible biomarker profiles. Certain AT(N) profiles indicate increased likelihood that LATE-NC might be present. The ‘N’ in AT(N) is in parentheses to indicate that it represents cumulative brain injury/neurodegeneration from all aetiologies and is not specific for any one aetiology. An assumption is that in Alzheimer’s disease, neurodegeneration is associated with tauopathy, and therefore in an A+T−(N)+ subject, the N+ is likely due to a comorbid non-Alzheimer’s disease pathophysiological process(es). If (N)+ is ascertained by an imaging measure that captures neurodegeneration as reflected medial temporal atrophy or hypometabolism, then this implicates LATE (often with hippocampal sclerosis) as a likely non-Alzheimer’s disease comorbidity. Similar logic applies to subjects with an A−T−(N)+ profile, the N+ is presumably due to a non-Alzheimer’s disease pathological process(es), and if the (N)+ measure is hippocampal atrophy, or medial temporal hypometabolism, then LATE is implicated (Fig. 5).
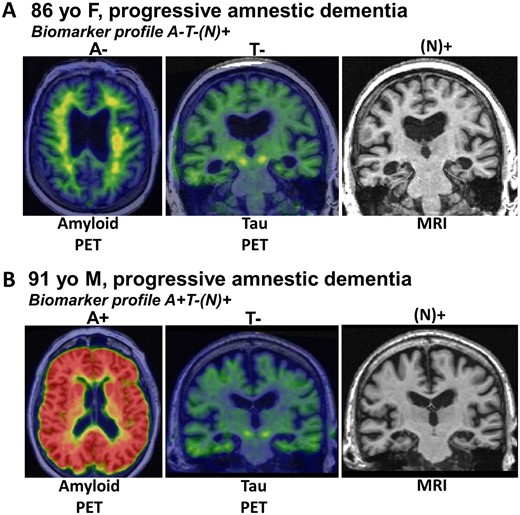
Biomarkers are currently not specific to LATE-NC. (A) Radiological scans from an 86-year-old female who suffered amnestic cognitive impairment compatible with ‘Probable Alzheimer’s disease’ diagnosis. However, the amyloid-β PET scan was negative, tau PET scan was also negative, and the MRI showed appreciable atrophy of the medial temporal lobes bilaterally. This combination is considered ‘A−T−N+’ and was diagnosed during life as ‘suspected non-Alzheimer’s pathology’ (SNAP). Autopsy within a year of the brain scans confirmed the presence of TDP-43 pathology and hippocampal sclerosis, which now is diagnosable as LATE-NC. (B) Another common biomarker combination, in the brain of a 91-year-old male with dementia. In this subject, the amyloid PET scan was positive, yet the tau PET scan was negative. The MRI again showed atrophy of the medial temporal lobes. The combination of pathologies—in this case presumed early ADNC and comorbid LATE-NC—is common, especially in the brains of subjects in advanced age.
SNAP is a non-specific biomarker-defined category that affects ∼15–30% of subjects in prior clinical series (Jack et al., 2012; Vos et al., 2013; Wisse et al., 2015; Burnham et al., 2016; Gordon et al., 2016), and includes a variety of non-Alzheimer’s disease aetiologies, but prominent among these is LATE. Autopsy studies indicate that LATE-NC can exist without other brain pathologies, but commonly co-occurs with ADNC (Jellinger, 2000; Attems and Jellinger, 2006; Josephs et al., 2014b, 2015, 2016); biomarker studies are consistent with those results. Botha et al. (2018) have shown that tau-PET-negative dementia can mimic Alzheimer’s disease clinically, suggesting that LATE is probably a common cause of tau-negative dementia. Further, a fluorodeoxyglucose (FDG) PET measure (the ratio of inferior to medial temporal metabolism) was elevated in autopsy proven LATE with hippocampal sclerosis compared to autopsy proven Alzheimer’s disease cases lacking LATE-NC (Botha et al., 2018). LATE-NC was confirmed at autopsy in two subjects with tau-PET-negative scans who both had elevated inferior to medial temporal FDG metabolism ratios. These data are compatible with the hypothesis that cognitively impaired tau-PET-negative subjects with marked medial temporal hypometabolism are likely to have LATE-NC. Other studies correlating autopsy findings with post-mortem magnetic resonance measures of regional tissue T2 relaxation times (Dawe et al., 2014), hippocampal shape (Dawe et al., 2011), and regional cortical volume measures (Kotrotsou et al., 2015) indicate that neuroimaging signatures of ADNC and LATE-NC may differ (see above). The shape differences in the medial temporal lobes associated with LATE-NC versus ADNC provide potential imaging biomarkers of LATE (Dawe et al., 2011; Makkinejad et al., 2019), whereas the evaluation of multiple brain regions is warranted as demonstrated in Fig. 3.
A non-specific biomarker of cumulative brain injury (N) may be useful in the context of LATE. If a biomarker for LATE is not forthcoming, then a quantitative in vivo indicator might remain the difference between the magnitude of an observed (N) biomarker minus the predicted (N) value given the results of all other known biomarkers. A predictive (but not diagnostic) LATE measure could be envisioned as the ‘residual of the regression’ of expected on observed medial/basal temporal neurodegeneration, given all knowable information about other pathological processes. Recent studies indicate that neurofilament light chain (NfL) might be a preferred biofluid (N) biomarker (Zetterberg, 2016; Kortvelyessy et al., 2018) but perturbation of NfL in LATE (plasma or CSF) remains to be tested. Moreover, elevated levels of NfL occur in many different causes of brain injury so NfL lacks disease specificity.
Although no specific LATE biomarker exists at present, the AT(N) system was designed explicitly to enable expansion to incorporate new biomarkers in categories beyond AT(N) (Jack et al., 2018). If or when a biomarker of LATE is validated, AT(N) could be expanded to ATL(N), where ‘L’ stands for LATE. The ultimate objective would be comprehensive characterization of many relevant brain pathologies in vivo using combinations of biomarkers. Future diagnostic biomarkers may be less centred on ADNC, and able to incorporate the common combinations of diseases that occur in ageing brain better. This concept is illustrated in Supplementary Table 2.
If a specific biomarker for LATE is developed, this may complement ongoing efforts to develop an optimal neuropathological assessment. LATE-NC may in the future be assessed along three dimensions: stage (i.e. anatomical distribution); subtype (i.e. differing histopathological patterns in a given region); and grade (i.e. severity or pathological load). While systems for subtyping or grading LATE-NC have yet to be validated, these may in the future be useful for early diagnosis, improved clinical prognosis, and development of new strategies to treat or prevent the disease.
Implications for Alzheimer’s disease and LATE clinical trials
Formalization of LATE diagnostic criteria and increased awareness of this disease should help guide the design and interpretation of Alzheimer’s disease clinical trials. Comorbid ADNC and LATE-NC becomes increasingly more prevalent with advancing age, and the mechanisms underlying each of these common lesions have independent effects on cognitive performance (Nelson et al., 2010). LATE-NC, when coexisting with ADNC, will have the potential to obscure the effects of a potential disease modifying agent on cognitive assessment results in living subjects. The primary outcome measures in disease-modifying Alzheimer’s disease clinical trials will remain cognitive or functional scales for the foreseeable future (Cummings et al., 2016; Register, 2018). Thus, the presence of LATE-NC will complicate interpretation of Alzheimer’s disease-specific treatment effects that are inferred from observed cognitive outcomes. Until there are biomarkers for LATE, clinical trials should be powered to account for TDP-43 proteinopathy.
LATE is among the common age-related diseases that can mimic the amnestic presentation of Alzheimer’s disease (Nelson et al., 2013), and it is one of many reasons why biological rather than clinical disease definitions are important in the era of disease modifying clinical trials (Jack et al., 2018). Biomarkers have roles for both inclusion and exclusion. It will be important, at recruitment of subjects into future disease-modifying Alzheimer’s disease clinical trials, to stratify according to major known predictors, including clinical features, genetics, and known biomarkers. This stratification will enable enrichment for subjects on the ADNC continuum (Sevigny et al., 2016) while excluding subjects likely to have high risk for LATE-NC (Botha et al., 2018). Even with best efforts at baseline, the multiplicity of diseases that occur in brains of older subjects will still require analyses according to subgroups. This is another reason why clinical trials in dementing diseases of ageing will require large sample sizes.
Research into Alzheimer’s disease has provided additional topical caveats (Gulisano et al., 2018; Hunter et al., 2018; Morris et al., 2018). For example, there is a danger that we fundamentally misunderstand the nature and complexity of processes related to TDP-43 proteinopathy, and this could lead to significant biases in the ways that we approach clinical diagnosis and clinical trials of LATE. For now, as with Alzheimer’s disease, the misfolded proteins provide a disease marker and a potential target for therapies.
Clinical trials directed at preventing or treating LATE, in isolation or in concert with other brain diseases, should be a major direction for future research. Performing such trials optimally will first require development of a specific LATE biomarker. For now, five alternative, but not mutually exclusive, approaches exist for developing disease-modifying therapies: (i) focus on pathways and gene products such as APOE ɛ4 that seem to be in common between Alzheimer’s disease, Lewy body disease, and LATE; (ii) focus on pathways and gene products such as TMEM106B and GRN that are shared between FTLD-TDP and LATE; (iii) focus on pathways and gene products such as ABCC9 and KCNMB2 that have been implicated by GWAS; (iv) focus on potential research subjects with the A−T−(N+) biomarker profile, who are now excluded from many Alzheimer’s disease-related clinical trials; and/or (v) focus on strategies to eliminate TDP-43 aggregates or to prevent the formation of these aggregates.
Conclusions and future directions
A key goal of this working group effort was to catalyse future research on LATE, an under-recognized condition that affects many older subjects. It is important to promote awareness in multiple scientific areas and to focus on translational and interdisciplinary approaches.
Development of specific LATE biomarker(s) should be a high scientific priority. While a sensitive and specific biomarker using neuroimaging or biofluids would be ideal, other disease markers could capitalize on existing metrics such as the AT(N) research guidelines with or without imaging or biofluid risk profiling. Developing biomarkers or other criteria to identify subjects with LATE would augment observational studies that seek to unravel the natural history of LATE, and its coevolution with other ageing-related diseases. With sufficient longitudinal observations, cause and effect inferences may become possible, and clinical trials implemented.
Further pathology studies will also be necessary. The consensus pathological classification scheme that we propose should be considered preliminary because much remains to be learned about LATE. The application of pathological subtyping has been useful in the context of FTLD-TDP (Lee et al., 2017; Mackenzie and Neumann, 2017; Pottier et al., 2018), and pathological subtyping may help refine LATE-NC endophenotypes for diagnostic and genetic studies (Josephs et al., 2019). At this point, there is no consensus about how or whether to apply such criteria for LATE-NC. A detailed characterization of the molecular pathology of TDP-43 is required for different cell types across brain regions in large population-representative samples. This should include characterization of various phosphorylation states, cleavage fragments, and other post-translational modifications of TDP-43. Further, each anti-TDP-43 antibody used should be assessed for potential cross-reactivity with other proteins or LATE-NC features in situ. It will also be important to determine the prevalence of all co-pathologies associated with LATE-NC, the impact of the molecular conformations and modifications of TDP-43, the cellular types involved, and the natural history of the disease. These advances will also assist in developing animal models.
Additional epidemiological, clinical, neuroimaging, and genetic studies will be important to better characterize the public health impact and clinical phenotypes for LATE. Further, LATE must be studied in more diverse populations and cohorts. Careful clinical assessments over time and into the oldest age groups is essential, along with detailed biological measures and autopsy, so that the complexity of ageing changes can be assessed (Brayne, 1993). In vivo and ex vivo imaging studies to determine the focal and more diffuse changes in the brains of subjects with LATE will also be important. Future studies may generate better insights into the clinical indices and cognitive features that are associated with increased probability of LATE-NC. Risk factors, protective influences, and other correlates could thus be identified to help prevent or predict LATE. For example, autoimmune disease may play a role in TDP-43 proteinopathy and LATE-NC in particular (Miller et al., 2013; Trieu et al., 2018). Optimally, future studies will complement traditional GWAS and gene-focused analyses with multi-omics studies to capture a greater appreciation of the complex mechanisms and diagnostic or therapeutic opportunities in the study of LATE.
Animal models and basic science research into LATE are imperative, with the caveat that the aged human brain is challenging to model accurately. Functional studies, including transmission animal models that use TDP-43 fibrils (Porta et al., 2018) or extracts from brains with LATE-NC injected into animals or cell cultures (Laferriere et al., 2019), can be combined with genetic studies to test hypotheses and to add statistical power for preclinical and hypothesis-testing experiments. Molecular studies that focus on TDP-43 and the upstream triggers and downstream molecular consequences are necessary to elucidate mechanisms of disease. Models that account for co-pathologies are rare at present, but have the potential to be highly informative. Ultimately, it is hoped that these collective research efforts will one day result in successful preventative and therapeutic strategies.
Abbreviations
- ADNC
Alzheimer’s disease neuropathological changes
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia (clinical syndrome)
- FTLD-TDP
frontotemporal lobar degeneration with TDP-43 proteinopathy
- GWAS
genome wide association study
- LATE-NC
limbic-predominant age-related TDP-43 encephalopathy neuropathological changes
- PPA
primary progressive aphasia
- SNAP
suspected non-Alzheimer (non-amyloid-β) pathophysiology (by biomarkers)
Acknowledgements
We sincerely thank the research volunteers, clinicians, staff, and other colleagues for their hard work. The LATE working group meeting organizational committee consisted of Drs Peter Nelson, Nina Silverberg, Dennis Dickson, Julie Schneider, John Trojanowski, and Helena Chui. The working group in-person meeting (October 17–18 in Atlanta, GA, USA) included most of the co-authors, and Drs Nina Silverberg, Eliezer Masliah, Cerise Elliott, and Linda Van Eldik attended and contributed. Statistical analyses in Figs 2 and 3 were performed by co-author Dr Lei Yu. Thanks to Dr Hannah Keage at the University of South Australia for editorial help.
Funding
Direct support for meeting logistics was provided via a NIA/NACC grant (supplemental to the parent NIH grant U01 AG016976 to W.A.K.). Support from the National Health and Medical Research Council of Australia (NHMRC) included NHMRC Senior Principal Research Fellowship (1079679) and other NHMRC grants (1132524, 1095127, 1037746) to G.H. Grant support from U.S. National Institutes of Health included grants P01 AG003949 (D.W.D.), R01 AG037491 (D.W.D.), P50 AG016574 (D.W.D.), R01 AG054449 (M.E.M.), P30 AG028303 (P.T.N. and G.A.J.), P30 AG012300 (C.L.W.), P30 AG049638 (T.J.M.), P30 AG010124 (J.Q.T.), P30 AG010161 (K.A., J.A.S., P.A.B.), P50 AG047366 (T.J.M.), P50 AG025688 (A.I.L.), P50 AG005131 (R.A.R.), R37 AG011378 (C.R.J.), R01 AG041851 (C.R.J.), R01 AG042210 (J.A.S.), R01 AG017917 (J.A.S.), R01 AG034374 (P.A.B.), UF1 AG053983 (T.J.M.), UF1 AG057707 (T.J.M.). UK grants included ARUK-PhD2014–19, and NIHR Senior Investigators award Ref: 534906 NF-SI-0616–10090 to co-author C.B. National Institute for Health Research, Senior Investigator Award, was awarded to C.B. for C.B. and S.H. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. S.H. is supported by the Addenbrooke’s Charitable Trust, the Paul G. Allen Family Foundation and Alzheimer’s Research, UK. S.R.K.H. is supported by Alzheimer’s Research, UK. See Appendix 1 for additional acknowledgments.
Competing interests
The authors report no competing interests.
Appendix 1
NACC data
We are extremely grateful to the many patients, clinicians, and other colleagues, who have worked so hard to provide and organize these data. The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG005131 (PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
References
Neuropathology Group.
Porta M.
Register OotF.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}