




Abstract
Nine neurodegenerative diseases, collectively referred to as polyglutamine (polyQ) diseases, are caused by expansion of a coding CAG DNA trinucleotide repeat. PolyQ diseases show a strong inverse correlation between CAG repeat length and age of disease onset (AO). Despite this, individuals with identical repeat expansion alleles can have highly variable disease onset indicating that other factors also influence AO. We examined AO in 148 individuals in 57 sibships from the SCA2 founder population in Cuba. The mutant CAG repeat allele explained 57% of AO variance. To estimate heritability of the residual variance after correction for SCA2 repeat length, we applied variance component analysis and determined the coefficient of intraclass correlation. We found that 55% of the residual AO variance was familial. To test candidate modifier alleles in this population, we selected 64 unrelated individuals from a set of 394 individuals who were highly discordant for AO after correction for SCA2 CAG repeat length. We hypothesized that long normal alleles in the other 8 polyQ disease genes were associated with premature disease onset in SCA2. Of the 8 genes tested, only long normal CAG repeats in the CACNA1A gene were associated with disease onset earlier than expected based on SCA2 CAG repeat size using non-parametric tests for alleles (P < 0.04) and genotypes (P < 0.023) after correction for multiple comparisons. CACNA1A variation explained 5.8% of the residual variation in AO. The CACNA1A calcium channel subunit represents an excellent candidate as a modifier of disease in SCA2. It is highly expressed in Purkinje cells (PCs) and is essential for the generation of the P/Q current and the complex spike in PCs. In contrast to other polyQ proteins, which are nuclear, the CACNA1A and SCA2 proteins are both cytoplasmic. Furthermore, small pathologic expansions of the polyQ domain in the CACNA1A protein lead to PC degeneration in SCA6. Future studies are needed to determine whether the modifier effect of CACNA1A relates to neuronal dysfunction or cell death of Purkinje neurons.
Introduction
A number of human genetic neurodegenerative diseases are caused by the expansion of a CAG repeat in the coding region of the respective gene. These mutations result in the expansion of a polyglutamine (polyQ) domain above a pathological threshold in the respective proteins, and these disorders have, therefore, been grouped under the term polyQ diseases. Common to all polyQ diseases is a significant inverse correlation between repeat length and age of onset, albeit with tremendous variability within each repeat length (reviewed in Van de Warrenburg et al., 2002, 2005; Pulst, 2003).
This variation suggests that there are a number of cis- and trans-acting genetic factors, non-allelic genetic modifiers, stochastic and environmental factors influencing age of onset (AO) in addition to the pathogenic allele itself. Although genetic modifiers have been identified in cell culture and in model systems, relatively little work has been done on humans defining factors that modify AO or even estimating the amount of AO variance attributable to shared genetic and environmental factors (familiality).
Understanding these factors is important for it may have important implications for understanding pathogenesis and improved counseling of presymptomatic individuals. It is conceivable that the modifying factors are more amenable to therapy than the causative mutation itself. Defining genetic modifiers will also facilitate the identification of environmental effects on the phenotype. Furthermore, stratification of patient cohorts by modifying factors will aid clinical trials by increasing homogeneity of patient groups.
SCA2 is caused by the expansion of a CAG repeat in the coding region of the ataxin-2 gene to more than 31 repeats. It is relatively rare worldwide, but common in the eastern part of Cuba with a prevalence of 50 patients per 100 000 (Hernandez et al., 1995; reviewed in Pulst, 2003). This founder population provides an unusual resource for genetic studies similar to the HD population in Venezuela (Wexler et al., 2004).
We examined the correlation of SCA2 age of onset and CAG repeat length in this population and found that a significant amount of AO variability was not determined by the mutant CAG allele. Using a strategy of allelic association in individuals highly discordant for age of onset, we identified alleles of the CACNA1A calcium channel subunit as modifiers of AO in SCA2 patients.
Methods
Patient population
Patients were recruited from the SCA2 population of the Ataxia Center in Holguin province, Cuba. Informed consent was obtained and all studies were approved by the appropriate review boards in Holguin and Los Angeles. For the analysis of residual variance and heritability, information from 148 SCA2 patients from 57 sibships was used. For the analysis of modifier genes, a set of 394 unrelated SCA2 patients were evaluated to select unrelated patients with highly discordant AO after adjustment for SCA2 repeat length. Individuals who were known to be related, including second cousins, were excluded from the analysis. We identified 30 patients who had an age of onset at least one standard deviation earlier than the mean adjusted on CAG repeats; 34 patients had disease onset at least one standard deviation later than the mean expected for CAG repeat length.
Determination of age of onset
Age of onset was considered the first sign of ataxia usually manifested by unsteadiness of gait. Whenever possible, age of onset was corroborated by other family members and could usually be related to specific handicaps such as inability to ride a bike, carry a child or handle tools.
DNA analysis
Blood samples were obtained and DNAs extracted from patient venous blood. Primers used to amplify CAG repeats and annealing temperatures are listed in Table 1. All repeat sizes were compared with standards, in which repeat size had been confirmed by DNA sequence analysis (Pulst et al., 1996).
Oligonucleotide primers
| Gene | Primer name | Primer sequence | Annealing temperature |
|---|---|---|---|
| SCA1 | Rep 1 | aactggaaatgtggacgtac | 60°C |
| Rep 2 | caacatgggcagtctgag | ||
| SCA2 | SCA2 A | gggcccctcaccatgtcg | 60°C |
| SCA2 B | cgggcttgcggacattgg | ||
| SCA3/MJD | MJD 25 | aagtgtaggtacactttccggt | 60°C |
| MJD 52 | ccagtgactactttgattcg | ||
| SCA7 | SCA7 C | tgttacattgtaggagcggaa | 57°C |
| SCA7 D | cacgactgtcccagcatcactt | ||
| SCA6 | SCA6 F | cacgtgtcctattcccctgtgatcc | 60°C |
| SCA6 R | tgggtacctccgagggccgctggtg | ||
| DRPLA | B37 F | caccagtctcaacacagcaccatc | 60°C |
| B37 R | cctccagtgggtggggaaatgctc | ||
| SCA17 | SCA17 A | Gaccccacagcctattcaga | 57°C |
| SCA17 B | Ttgactgctgaacggctgca | ||
| SBMA | SBMA F | tccagaatctgttccagagcgtgc | 57°C |
| SBMA R | gctgtgaaggttgctgttcctcat | ||
| HD | HD 1 | ggcggctgaggaagctgagga | 60°C |
| HD 3 | ggcggtggcggctgttgctgctgctgctgc | ||
| hSKCa3 | Europe F | Aagtgcccctgtccatcctct | 60°C |
| Europe R | Gccaagcaagtggtcattgag |
| Gene | Primer name | Primer sequence | Annealing temperature |
|---|---|---|---|
| SCA1 | Rep 1 | aactggaaatgtggacgtac | 60°C |
| Rep 2 | caacatgggcagtctgag | ||
| SCA2 | SCA2 A | gggcccctcaccatgtcg | 60°C |
| SCA2 B | cgggcttgcggacattgg | ||
| SCA3/MJD | MJD 25 | aagtgtaggtacactttccggt | 60°C |
| MJD 52 | ccagtgactactttgattcg | ||
| SCA7 | SCA7 C | tgttacattgtaggagcggaa | 57°C |
| SCA7 D | cacgactgtcccagcatcactt | ||
| SCA6 | SCA6 F | cacgtgtcctattcccctgtgatcc | 60°C |
| SCA6 R | tgggtacctccgagggccgctggtg | ||
| DRPLA | B37 F | caccagtctcaacacagcaccatc | 60°C |
| B37 R | cctccagtgggtggggaaatgctc | ||
| SCA17 | SCA17 A | Gaccccacagcctattcaga | 57°C |
| SCA17 B | Ttgactgctgaacggctgca | ||
| SBMA | SBMA F | tccagaatctgttccagagcgtgc | 57°C |
| SBMA R | gctgtgaaggttgctgttcctcat | ||
| HD | HD 1 | ggcggctgaggaagctgagga | 60°C |
| HD 3 | ggcggtggcggctgttgctgctgctgctgc | ||
| hSKCa3 | Europe F | Aagtgcccctgtccatcctct | 60°C |
| Europe R | Gccaagcaagtggtcattgag |
Oligonucleotide primers
| Gene | Primer name | Primer sequence | Annealing temperature |
|---|---|---|---|
| SCA1 | Rep 1 | aactggaaatgtggacgtac | 60°C |
| Rep 2 | caacatgggcagtctgag | ||
| SCA2 | SCA2 A | gggcccctcaccatgtcg | 60°C |
| SCA2 B | cgggcttgcggacattgg | ||
| SCA3/MJD | MJD 25 | aagtgtaggtacactttccggt | 60°C |
| MJD 52 | ccagtgactactttgattcg | ||
| SCA7 | SCA7 C | tgttacattgtaggagcggaa | 57°C |
| SCA7 D | cacgactgtcccagcatcactt | ||
| SCA6 | SCA6 F | cacgtgtcctattcccctgtgatcc | 60°C |
| SCA6 R | tgggtacctccgagggccgctggtg | ||
| DRPLA | B37 F | caccagtctcaacacagcaccatc | 60°C |
| B37 R | cctccagtgggtggggaaatgctc | ||
| SCA17 | SCA17 A | Gaccccacagcctattcaga | 57°C |
| SCA17 B | Ttgactgctgaacggctgca | ||
| SBMA | SBMA F | tccagaatctgttccagagcgtgc | 57°C |
| SBMA R | gctgtgaaggttgctgttcctcat | ||
| HD | HD 1 | ggcggctgaggaagctgagga | 60°C |
| HD 3 | ggcggtggcggctgttgctgctgctgctgc | ||
| hSKCa3 | Europe F | Aagtgcccctgtccatcctct | 60°C |
| Europe R | Gccaagcaagtggtcattgag |
| Gene | Primer name | Primer sequence | Annealing temperature |
|---|---|---|---|
| SCA1 | Rep 1 | aactggaaatgtggacgtac | 60°C |
| Rep 2 | caacatgggcagtctgag | ||
| SCA2 | SCA2 A | gggcccctcaccatgtcg | 60°C |
| SCA2 B | cgggcttgcggacattgg | ||
| SCA3/MJD | MJD 25 | aagtgtaggtacactttccggt | 60°C |
| MJD 52 | ccagtgactactttgattcg | ||
| SCA7 | SCA7 C | tgttacattgtaggagcggaa | 57°C |
| SCA7 D | cacgactgtcccagcatcactt | ||
| SCA6 | SCA6 F | cacgtgtcctattcccctgtgatcc | 60°C |
| SCA6 R | tgggtacctccgagggccgctggtg | ||
| DRPLA | B37 F | caccagtctcaacacagcaccatc | 60°C |
| B37 R | cctccagtgggtggggaaatgctc | ||
| SCA17 | SCA17 A | Gaccccacagcctattcaga | 57°C |
| SCA17 B | Ttgactgctgaacggctgca | ||
| SBMA | SBMA F | tccagaatctgttccagagcgtgc | 57°C |
| SBMA R | gctgtgaaggttgctgttcctcat | ||
| HD | HD 1 | ggcggctgaggaagctgagga | 60°C |
| HD 3 | ggcggtggcggctgttgctgctgctgctgc | ||
| hSKCa3 | Europe F | Aagtgcccctgtccatcctct | 60°C |
| Europe R | Gccaagcaagtggtcattgag |
Statistical analysis
Regression analysis was performed to fit the relationship between age of onset and the repeat length in 148 SCA2 patients. The SCA2 repeat length was used as the independent variable. Both age of onset and logarithmically transformed age of onset were examined as dependent variables. Use of the logarithmically transformed age of onset yielded a slightly better fit.
Calculation of residual age of onset variance heritability
One-way ANOVA was used to access the variance components of the age of onset adjusted by SCA2 repeat length in 148 individuals from 57 sibships. The variance component among the sibships and the error variance within the sibships were used to calculate the coefficient of intraclass correlation (Sokal and Rohlff, 1997). The estimated intraclass correlation is an upper bound of heritability for the residual age of onset.
Test of association between polyQ genes and age of onset
We selected two groups of patients from two tails of the residual age of onset distribution. The first group (premature onset, n = 30) consisted of unrelated patients who had an age of onset one standard deviation earlier than the mean age of onset. The second group (delayed onset, n = 34) had an age of onset one standard deviation later than the mean. As CAG repeat alleles are not normally distributed and often show a bi- or trimodal distribution (see Fig. 2), we chose the non-parametric Mann–Whitney rank test (U-test), although these tests usually have reduced power to detect differences compared with parametric tests. All statistical analyses were performed using SAS version 8.2. A regression model was used to estimate the percent of variance that was explained by the CACNA1A CAG repeat.
Results
Correlation of age of onset and CAG repeat length in Cuban SCA2
We examined age of onset and SCA2 CAG repeat length in 148 Cuban SCA2 patients. These patients derive from a founder population that migrated to the eastern part of Cuba in several waves of immigration during the last 300 years. All affected patients share the same chromosomal haplotype surrounding the mutant CAG repeat consistent with the presence of a founder population (Hernandez et al., 1995). The sibships were drawn from a larger cohort of 394 individuals for whom age of onset and SCA2 CAG repeat length were known.
Mean age of onset and mean length of the expanded CAG repeat allele were identical for the total cohort and the smaller sibship cohort (Table 2). There was no significant difference in the age of onset between males (31.7 ± 13.9) and females (32.8 ± 13.3). Means of CAG repeat length for both genders were virtually identical (males: 40.5 ± 4.5; females 40.3 ± 3.8).
Characteristics of SCA2 cohorts
| Cohort | Number of individuals | Number of females | Age of onset | Mutant CAG repeat | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | |||||
| Total | 394 | 167 | 32.2 ± 13.7 | 30 | 40.4 ± 4.1 | 40 | ||
| Sibships | 148 | 71 | 33.7 ± 13.7 | 32 | 39.8 ± 3.2 | 39 | ||
| Premature | 30 | 13 | 23.1 ± 8.12 | 20 | 39.7 ± 2.2 | 40 | ||
| Delayed | 34 | 12 | 40.7 ± 11.9 | 40 | 40.1 ± 2.9 | 40 | ||
| Cohort | Number of individuals | Number of females | Age of onset | Mutant CAG repeat | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | |||||
| Total | 394 | 167 | 32.2 ± 13.7 | 30 | 40.4 ± 4.1 | 40 | ||
| Sibships | 148 | 71 | 33.7 ± 13.7 | 32 | 39.8 ± 3.2 | 39 | ||
| Premature | 30 | 13 | 23.1 ± 8.12 | 20 | 39.7 ± 2.2 | 40 | ||
| Delayed | 34 | 12 | 40.7 ± 11.9 | 40 | 40.1 ± 2.9 | 40 | ||
Characteristics of SCA2 cohorts
| Cohort | Number of individuals | Number of females | Age of onset | Mutant CAG repeat | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | |||||
| Total | 394 | 167 | 32.2 ± 13.7 | 30 | 40.4 ± 4.1 | 40 | ||
| Sibships | 148 | 71 | 33.7 ± 13.7 | 32 | 39.8 ± 3.2 | 39 | ||
| Premature | 30 | 13 | 23.1 ± 8.12 | 20 | 39.7 ± 2.2 | 40 | ||
| Delayed | 34 | 12 | 40.7 ± 11.9 | 40 | 40.1 ± 2.9 | 40 | ||
| Cohort | Number of individuals | Number of females | Age of onset | Mutant CAG repeat | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | |||||
| Total | 394 | 167 | 32.2 ± 13.7 | 30 | 40.4 ± 4.1 | 40 | ||
| Sibships | 148 | 71 | 33.7 ± 13.7 | 32 | 39.8 ± 3.2 | 39 | ||
| Premature | 30 | 13 | 23.1 ± 8.12 | 20 | 39.7 ± 2.2 | 40 | ||
| Delayed | 34 | 12 | 40.7 ± 11.9 | 40 | 40.1 ± 2.9 | 40 | ||
Using a logarithmic fit, a significant inverse correlation was found between AO and repeat length (r2 = 0.57, P < 0.0001). This indicated that the SCA2 CAG repeat accounted for slightly <60% of the age of onset variance in SCA2 and that factors other than the mutant allele accounted for 40% of the residual variance.
Heritability of residual age of onset variance
To determine familiality or an upper bound for heritability of the residual variance (adjusted AO), we analysed age of onset in 57 sibships comprising a total of 148 individuals. Using variance-components analysis we estimated the between and within sibling pair variances of residual AO variance allowing for the computation of the sibling intraclass correlation. Sibling intraclass correlation was 0.274 indicating an upper bound for heritability of ∼55%. This value is close to heritability estimates for residual AO variance in Huntington disease (Li et al., 2003; Wexler et al., 2004) and indicates a significant genetic and shared familial environmental contribution to AO in SCA2.
Genetic modifiers of age of onset
To begin the search for modifier genes, we examined one set of candidates: the other expanded CAG repeat disease genes. We used data from 394 Cuban SCA2 patients to select unrelated patients with highly discordant adjusted AO (Fig. 1). The first group (premature onset, n = 30) consisted of patients who had an age of onset one standard deviation earlier than the mean AO adjusted on CAG repeats. The second group (delayed onset, n = 34) had disease onset one standard deviation later than the mean. The two groups did not differ in the means of the mutant CAG repeat length (Table 2). As expected, the age of onset in the two groups was significantly different with a difference in the mean ages of 17.6 years. This confirmed that these groups represented highly discordant phenotypes with regard to residual AO (after correction for SCA2 CAG repeat length).
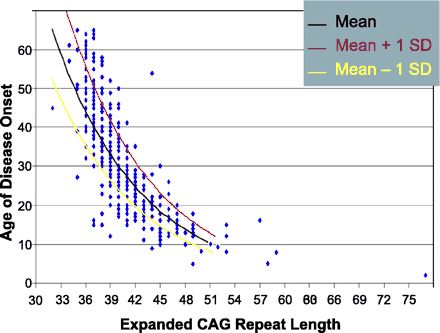
Scatterplot of SCA2 CAG repeat length and age of onset in Cuban SCA2 patients; red and yellow lines denote one standard deviation boundaries for age of onset.
We determined the size of CAG repeat alleles in the SCA1, SCA3, CACNA1A (SCA6), SCA7, SCA17, huntingtin, DRPLA, and androgen receptor genes in the two patient groups and tested the hypothesis that larger normal alleles in these genes predisposed to earlier disease onset. A priori, several of these genes were less likely to be good candidates. The androgen receptor is mainly expressed in spinal motor neurons and expansion of the CAG repeat causes spinobulbar muscular atrophy without cerebellar involvement (Adachi et al., 2005). Similarly, HD affects primarily cortical and basal ganglia neurons, although ataxia is observed in juvenile HD and Purkinje cell (PC) number is decreased in adult patients with HD (Jeste et al., 1984). No correlation between the CAG repeats in these 8 genes and the pathologic SCA2 allele was detected confirming that CAG expansions at different genomic locations are independent events (data not shown).
We examined the frequency of the longest allele for each respective polyQ gene in the premature and delayed onset groups. We also compared the frequencies of CAG genotypes. The CAG genotype for respective diseases was determined by summing the CAG repeats on both alleles (Fig. 2; Table 3). Except for the CACNA1A gene, no difference in CAG repeat number was detected between the premature and the delayed onset group for all genes examined. A distribution typical for 7 of the 8 genes is exemplified by DRPLA CAG repeats. CAG repeat numbers in the premature and delayed onset group are virtually identical both for the larger DRPLA allele and for the DRPLA genotype (Fig. 2A and B). As is typical for most polyQ disease genes, CAG repeats were not normally distributed and showed a bi- or trimodal distribution. The lack of a normal distribution necessitated the use of the non-parametric Mann–Whitney U-test test for statistical analyses. Significance levels uncorrected for multiple comparisons for all genes are shown in Table 3.
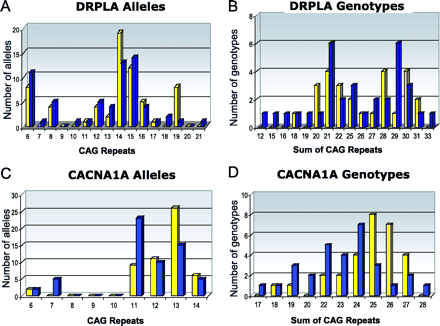
Distribution of alleles and genotypes for CAG repeats in the DRPLA and CACNA1A genes in the premature (yellow) and delayed (blue) onset groups. Long CACNA1A alleles and genotypes are more common in the premature onset group. (A) DRPLA alleles; (B) DRPLA genotypes; (C) CACNA1A alleles; (D) CACNA1A genotypes. Genotypes are determined by summing the CAG repeats in each allele.
Analysis of allele and genotype distribution
| Gene | Disease | Mann–Whitney significance level | ||
|---|---|---|---|---|
| For largest allele | For genotype | |||
| Ataxin-1 | SCA1 | P = 0.0846 | P = 0.0891 | |
| Ataxin-3 | SCA3 | P = 0.2309 | P = 0.2043 | |
| CACNA1A | SCA6 | P = 0.0050 | P = 0.0029 | |
| Ataxin-7 | SCA7 | P = 0.9420 | P = 0.9543 | |
| TBP | SCA17 | P = 0.6567 | P = 0.6478 | |
| Atrophin-1 | DRPLA | P = 0.1136 | P = 0.1215 | |
| AR | SBMA | P = 0.6241 | P = 0.6202 | |
| Huntingtin | HD | P = 0.8490 | P = 0.9216 | |
| Gene | Disease | Mann–Whitney significance level | ||
|---|---|---|---|---|
| For largest allele | For genotype | |||
| Ataxin-1 | SCA1 | P = 0.0846 | P = 0.0891 | |
| Ataxin-3 | SCA3 | P = 0.2309 | P = 0.2043 | |
| CACNA1A | SCA6 | P = 0.0050 | P = 0.0029 | |
| Ataxin-7 | SCA7 | P = 0.9420 | P = 0.9543 | |
| TBP | SCA17 | P = 0.6567 | P = 0.6478 | |
| Atrophin-1 | DRPLA | P = 0.1136 | P = 0.1215 | |
| AR | SBMA | P = 0.6241 | P = 0.6202 | |
| Huntingtin | HD | P = 0.8490 | P = 0.9216 | |
AR, androgen receptor; DRPLA, dentatorubral-pallidoluysian atrophy; SBMA, spinobulbar muscular atrophy. HD, Huntington disease.
Analysis of allele and genotype distribution
| Gene | Disease | Mann–Whitney significance level | ||
|---|---|---|---|---|
| For largest allele | For genotype | |||
| Ataxin-1 | SCA1 | P = 0.0846 | P = 0.0891 | |
| Ataxin-3 | SCA3 | P = 0.2309 | P = 0.2043 | |
| CACNA1A | SCA6 | P = 0.0050 | P = 0.0029 | |
| Ataxin-7 | SCA7 | P = 0.9420 | P = 0.9543 | |
| TBP | SCA17 | P = 0.6567 | P = 0.6478 | |
| Atrophin-1 | DRPLA | P = 0.1136 | P = 0.1215 | |
| AR | SBMA | P = 0.6241 | P = 0.6202 | |
| Huntingtin | HD | P = 0.8490 | P = 0.9216 | |
| Gene | Disease | Mann–Whitney significance level | ||
|---|---|---|---|---|
| For largest allele | For genotype | |||
| Ataxin-1 | SCA1 | P = 0.0846 | P = 0.0891 | |
| Ataxin-3 | SCA3 | P = 0.2309 | P = 0.2043 | |
| CACNA1A | SCA6 | P = 0.0050 | P = 0.0029 | |
| Ataxin-7 | SCA7 | P = 0.9420 | P = 0.9543 | |
| TBP | SCA17 | P = 0.6567 | P = 0.6478 | |
| Atrophin-1 | DRPLA | P = 0.1136 | P = 0.1215 | |
| AR | SBMA | P = 0.6241 | P = 0.6202 | |
| Huntingtin | HD | P = 0.8490 | P = 0.9216 | |
AR, androgen receptor; DRPLA, dentatorubral-pallidoluysian atrophy; SBMA, spinobulbar muscular atrophy. HD, Huntington disease.
In contrast to the other polyQ disease genes, CAG repeat length in the CACNA1A gene had a significant effect on AO in SCA2 (Fig. 2C and D). A higher frequency of longer CACNA1A alleles (P = 0.005) and genotypes (P = 0.0029) was found in the premature onset group (Fig. 2; Table 3). These results remained significant even after adjustment for multiple comparisons using the conservative Bonferroni correction (P = 0.040 for alleles; P = 0.023 for genotypes, adjusted for multiple comparisons).
In order to estimate the effect size of CACNA1A variants on AO in SCA2, we used a regression model incorporating SCA2 and CACNA1A repeat lengths into the analysis. The residual age of onset after adjusting for the SCA2 CAG repeat length was used as the dependent variable, the CACNA1A repeat as the independent variable. The P-value from the F-statistic was 0.055 and the estimated percentage of the residual AO variance explained by the CACNA1A genotype was 5.81%.
Discussion
SCA2 is a progressive neurodegenerative disease that typically and predominantly involves PCs, although other neuronal groups are involved as well. This is reflected in the phenotype that includes cerebellar ataxia, spasticity, neuropathy, dementia and parkinsonian features (Cancel et al., 1997; Geschwind et al., 1997; Schols et al., 1997). SCA2 shares with other polyQ diseases a strong inverse correlation between repeat length and age of onset. We chose to examine this relationship in the Cuban SCA2 founder population in the province of Holguin. The study of a founder population has certain inherent advantages and disadvantages. On the one hand, founder populations tend to be genetically more homogeneous and often also live in a more homogeneous environment. This may facilitate the detection of phenotypic modifiers. On the other, findings in founder populations may be difficult to generalize to other populations.
The Cuban SCA2 populations showed a strong inverse correlation between repeat length and age of onset. In other SCA2 populations, values for r2 have been reported that ranged from 0.47 to 0.8 and most commonly were ∼0.6 (Cancel et al., 1997; Geschwind et al., 1997; Riess et al., 1997; Giuffrida et al., 1999; Van de Warrenburg et al., 2002, 2005). The r2 of 0.57 observed in this study is in good agreement with studies in other ethnic and geographic SCA2 groups and agrees very closely with an r2 of 0.59 seen in Dutch SCA2 families (Van de Warrenburg et al., 2002). This supports the notion that the effect of the SCA2 CAG repeat on AO in the Cuban founder population does not significantly differ from other populations.
Despite this highly significant correlation, the observed values for r2 suggest that a significant amount of AO variance is not determined by the length of the mutant CAG repeat. Surprisingly little is known about heritability of this residual AO variance in dominant ataxias. Ranum et al. (1994) examined variability of AO between different SCA1 families and concluded that AO appeared to cluster within families suggesting effects of shared genetic and environmental background on AO. Two studies examined the contribution of the normal allele in SCA2 and found that it had either a small, but insignificant influence on AO (Van de Warrenburg et al., 2002) or no effect (Cancel et al., 1997). The normal allele in Cuban SCA2 is not highly variable; in fact, only two patients in this study had 23 SCA2 CAG repeats, the remainder had 22. Therefore, the effect of the normal SCA2 allele on AO could not be examined in our study.
No formal analysis of the residual AO variance in dominant ataxias has previously been reported. We used the coefficient of intraclass correlation in sibships to estimate heritability of the residual AO variance. Doubling this coefficient provides an upper bound for heritability (Djoussé et al., 2003). In human populations it is difficult to differentiate familiality, which combines shared genetic and shared environmental factors, from heritability in the strict sense, which denotes the proportion of phenotypic variance attributable to genetic variance. Our estimate that 55% of the residual AO variance in SCA2 is familial is in good agreement with studies in HD, another polyQ disease. Heritability (familiality) of residual AO variance in HD ranged from 38 to 56% (Djousse et al., 2003; Li et al., 2003; Wexler et al., 2004).
Given the phenotypic overlap between different polyQ diseases and the known functional effects of a pathologically expanded polyQ tract, we sought to determine whether variation of the CAG tract in 8 diseases known to cause neurodegenerative diseases influenced AO in SCA2. We hypothesized that longer normal CAG repeats in other polyQ disease genes would result in an earlier disease onset. We examined all known polyQ disease repeats, although some of them were unlikely candidates a priori. For example, the androgen receptor is not significantly expressed in PCs and ataxia is not part of the SBMA phenotype. Even SCA3, which shows significant ataxia, is predominated by degeneration of dentate neurons and not of PCs (Koeppen et al., 1999).
To maximize power, we chose a design that utilized SCA2 patients with highly discordant ages of disease onset after correction for the effect of the mutant SCA2 allele. Our premature onset and delayed onset groups of patients were separated by two standard deviations in their AO based on SCA2 CAG repeat length (Fig. 1). Although they had virtually identical mean SCA2 CAG repeat lengths, the mean age of disease onset in the two groups differed by 18 years (Table 2). Of note, the means of the mutant CAG length in the two groups were also identical with the mean CAG repeat length for the total sample of 394 individuals (Table 2).
We tested association with the longer CAG allele for each polyQ disease gene and with the genotype determined by summing CAG repeats in the two alleles. Of the 8 CAG repeats tested, only the CAG repeat in the CACNA1A calcium channel subunit gene showed a significant difference between the premature and delayed onset group (Fig. 2; Table 3). The association was observed both for the larger normal CACNA1A allele and the CACNA1A genotype and remained significant after correction for multiple comparisons using the conservative Bonferroni correction. Variation in the CACNA1A polyQ repeat accounted for ∼6% of the residual AO variance. This compares with an effect of ∼13% of variation in a TAA repeat in the GluR6 kainate receptor on AO in HD (Rubinsztein et al., 1997).
To our knowledge, no modifiers of AO have been described for any of the dominant ataxias except for one study of SCA2 using a large number of pedigrees from different ethnic backgrounds. This study did not find an association of the CACNA1A polyQ repeat on AO in SCA2 (Hayes et al., 2000). However, that study may have been underpowered to detect such an effect. Alternatively, the observed effect in the current study may be limited to the Cuban founder population.
As is common to all genetic association studies our findings cannot directly address a causative explanation of the observed effect of CACNA1A on age of onset in SCA2. Several explanatory models are consistent with our observations. First, the observed allelic association could be due to the effects of a linked functional polymorphism. A TAA repeat in the 3′ untranslated portion of the GluR6 kainate receptor gene is thought to be a neutral polymorphism in linkage disequilibrium with a functional variant (Rubinsztein et al., 1997). Although we cannot exclude this possibility on formal grounds, it is less likely as the CACNA1A CAG repeat is in the coding region of the gene. In addition, trinucleotide repeats show a higher mutation rate than bi-allelic single nucleotide polymorphisms, and would thus be less likely to be in linkage disequilibrium with an adjacent functional variant, which would make it more difficult to detect such an association.
On the other hand, CACNA1A itself is a plausible biological candidate as an AO modifier in ataxia for several reasons. The CACNA1A channel subunit is highly expressed in PCs and is responsible for the complex spike in PCs (Jun et al., 1999; Ishikawa et al., 1999). Proper functioning of the CACNA1A channel in PCs is important in PC function and survival. Expansions of the polyQ domain in CACNA1A cause the progressive neurological disease SCA6, and 19 glutamines already represent a pathologic repeat (Mariotti et al., 2001). Pathologic expansion of the polyQ domain in CACNA1A leads to altered channel activity, although different molecular mechanisms including abnormal activation and inactivation and current density have been implicated (reviewed in Gomez, 2001). Deficiency of CACNA1A, on the other hand, also leads to dysfunction, and in the mouse is associated with dystonia and ataxia accompanied by progressive loss of PCs (Jun et al., 1999; Fletcher et al., 2001).
Variation in the polyQ domain in CACNA1A could influence AO either by directly interacting with ataxin-2 or by altering channel function. It is likely that PCs expressing mutant ataxin-2 show increased sensitivity to subtle alterations in CACNA1A functioning that would not in themselves have an effect in otherwise normal individuals. Direct interaction of CACNA1A and mutant ataxin-2 could have several effects. First, longer polyQ domains could enhance initial steps in aggregation of ataxin-2 (Chen et al., 2002). Cytoplasmic aggregates have been observed in humans with SCA2 and in animals expressing mutant ataxin-2[Q58] (Huynh et al., 1999, 2000). Although aggregates are clearly associated with disease in humans and rodents, their direct pathogenic role has not been established in SCA2 (Huynh et al., 2003). Alternatively, mutant ataxin-2 aggregates could sequester CACNA1A protein resulting in reduced CACNA1A molecules at the cell surface and reduced P/Q currents.
Our preliminary immunofluorescent studies in SCA2 brains have demonstrated co-localization of CACNA1A protein in SCA2 aggregates. It is important to recognize, however, that post-mortem examination can only provide a glimpse at the late stages of the disease. Intercrosses of animal models expressing SCA2 transgenes and mutant Cacna1a will be best to address the role of this calcium channel in SCA2 and address the important question as to whether the CACNA1A modifier effect relates to neuronal dysfunction, to neuronal death or both.
In summary, our results indicate that genetic analysis of age of onset variance in SCA2 is feasible as about half of the residual AO variance is heritable. The heritable component of the residual age of onset variance contributes ∼20% to the overall AO variance. The analysis of candidate alleles using individuals with highly discordant phenotypes represents a powerful approach and should encourage genetic modifier studies in other SCA2 populations.
We thank our Cuban patients for participation in these investigations. This work was supported by grants from the National Institutes of Health (RO1 NS33123), the National Ataxia Foundation, and the Carmen and Louis Warschaw Endowment for Neurology (all to SMP).
References
Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients.
Cancel G, Dűrr A, Didierjean O, Imbert G, Burk K, Lezin A, et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families.
Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation.
Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, et al. Interaction of normal and expanded CAG repeat sizes influence age at onset of Huntington disease.
Fletcher CF, Tottene A, Lennon VA, Wislon SM, Dubel SJ, Paylor R, et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity.
Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 (SCA2) trinucleotide repeat in patients with autosomal dominant cerebellar ataxia.
Giuffrida F, Lanza S, Restivo DA, Saponara R, Valvo SC, Le Pira F, et al. Clinical and molecular analysis of 11 Sicilian SCA2 families: influence of gender on age of onset.
Hayes S, Turecki G, Brisebois K, Lopes-Cendes I, Gaspar C, Riess O, et al. CAG repeat length in RAI1 is associated with age at onset variability in spinocerebellar ataxia type 2 (SCA2).
Hernandez A, Magarino C, Gispert S, Santos N, Lunkes A, Orozco G, et al. Genetic mapping of the spinocerebellar ataxia 2 (SCA2) locus on chromosome 12q23-q24.1.
Huynh DH, Del Bigio MR, Sahba S, Ho DH, Pulst SM. Expression of ataxin 2 in brains from normal individuals and patients with Alzheimer disease and spinocerebellar ataxia 2 (SCA2).
Huynh DP, Figueroa K, Hoang N, Pulst SM. Nuclear localization or inclusion body formation are not necessary for SCA2 pathogenesis in mouse or human.
Huynh DG, Yang HT, Vakharia H, Nguyen D, Pulst SM. Expansion of the polyglutamine repeat in ataxin-2 causes its subcellular redistribution and apoptotic cell death.
Ishikawa K, Fujigasaki H, Saegusa H, Ohwada K, Fujita T, Iwamoto H, et al. Abundant expression and cytoplasmic aggregation of (alpha) 1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6.
Jeste DV, Barban L, Parisi J. Reduced Purkinje cell density in Huntington's disease.
Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the (1A)-subunit.
Koeppen AH, Dickson AC, Lamarche JB, Robitaille Y. Synapses in the hereditary ataxias.
Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dode C, et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study.
Mariotti C, Gellera C, Grisoli M, Mineri R, Castucci A, Di Donato S. Pathogenic effect of an intermediate-size SCA-6 allele (CAG)(19) in a homozygous patient.
Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Identification of the SCA2 gene: moderate expansion of a normally biallelic trinucleotide repeat.
Pulst SM. Spinocerebellar ataxia. In: Pulst SM, editor. Genetics of movement disorders. San Diego: Academic Press;
Ranum LP, Chung MY, Banfi S, Bryer A, Schut LJ, et al. Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age of onset.
Riess O, Laccone FA, Gispert S, Schols L, Zuhlke C, Viera-Saecker AM, et al. SCA2 trinucleotide expansion in German SCA patients.
Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, et al. Genotypes at the GluR6 kainate receptor locus are associated with variation in age of onset of Huntington disease.
Schols L, Gispert S, Vorgerd M, Menezes Viera-Saecker AM, Blanke P, Auburger G, et al. Spinocerebellar ataxia type 2: genotype and phenotype in German kindreds.
Van de Warrenburg BP, Sinke RJ, Verschuuren-Bemelmans CC, Scheffer H, Brunt ER, Ippel PF, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis.
Van de Warrenburg BP, Hendriks H, Dűrr A, van Zuijlen MC, Stevanin G, Camuzat A, et al. Age at onset variance analysis in spinocerebellar ataxias: a cohort study in a Dutch–French cohort.
Author notes
1Division of Neurology and Rose Moss Laboratory for Parkinson and Related Diseases, Burns and Allen Research Institute, Cedars-Sinai Medical Center, 2Department of Medicine, 3Department of Neurobiology, 4Department of Pediatrics and 5Department of Epidemiology, CSMC, Medical Genetics Institute, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA and 6Clínica para la Investigación y Rehabilitación de las Ataxias Hereditarias, Holguín, Cuba


{kind=link}
{kind=link}