




Abstract
Spinal and bulbar muscular atrophy (SBMA) is an adult-onset motoneuron disease caused by a CAG-repeat expansion in the androgen receptor (AR) gene and for which no curative therapy exists. However, since recent research may provide opportunities for medical treatment, information concerning the natural history of SBMA would be beneficial in planning future clinical trials. We investigated the natural course of SBMA as assessed by nine activities of daily living (ADL) milestones in 223 Japanese SBMA patients (mean age at data collection = 55.2 years; range = 30–87 years) followed from 1 to 20 years. All the patients were diagnosed by genetic analysis. Hand tremor was an early event that was noticed at a median age of 33 years. Muscular weakness occurred predominantly in the lower limbs, and was noticed at a median age of 44 years, followed by the requirement of a handrail to ascend stairs at 49, dysarthria at 50, dysphagia at 54, use of a cane at 59 and a wheelchair at 61 years. Twenty-one of the patients developed pneumonia at a median age of 62 and 15 of them died at a median age of 65 years. The most common cause of death in these cases was pneumonia and respiratory failure. The ages at onset of each ADL milestone were strongly correlated with the length of CAG repeats in the AR gene. However CAG-repeat length did not correlate with the time intervals between each ADL milestone, suggesting that although the onset age of each ADL milestone depends on the CAG-repeat length in the AR gene, the rate of disease progression does not. The levels of serum testosterone, an important triggering factor for polyglutamine-mediated motoneuron degeneration, were maintained at relatively high levels even at advanced ages. These results provide beneficial information for future clinical therapeutic trials, although further detailed prospective studies are also needed.
Abbreviations
- ADL
activities of daily living
- ALT
alanine aminotransferase
- AR
androgen receptor
- AST
aspartate aminotransferase
- CK
creatine kinase
- HbA1c
haemoglobin A1c
- SBMA
spinal and bulbar muscular atrophy
Introduction
Spinal and bulbar muscular atrophy (SBMA) is a neurodegenerative disorder of motoneurons characterized by proximal limb muscular atrophy, bulbar involvement, marked contraction fasciculation, hand tremor and gynaecomastia (Kennedy et al., 1968; Sobue et al., 1989). SBMA is caused by a CAG-repeat expansion in the first exon of the androgen receptor (AR) gene on the X-chromosome (La Spada et al., 1991). Similar to other triplet repeat diseases, the age at onset of disease has been inversely linked to the size of the CAG-repeat expansions (Andrew et al., 1993; Sasaki et al., 1996; Rosenblatt et al., 2003). For example, an association between the age at onset of limb muscle weakness and the CAG-repeat length has been demonstrated (Doyu et al., 1992; Igarashi et al., 1992; La Spada et al., 1992; Shimada et al., 1995; Sinnreich et al., 2004). Nuclear accumulation of mutant AR with expanded polyglutamines in motoneurons, as well as in other cells, has been shown to be a major pathogenic process (Li et al., 1998a, b; Adachi et al., 2005). However, the progression and prognosis of SBMA has not been assessed in detail, particularly concerning the influence of CAG-repeat size, the decline of the activities of daily living (ADL) with disease progression and the determination of functional prognosis. Some SBMA studies reported no correlation between the progression of the clinical course and the number of CAG repeats (Lund et al., 2001; Sperfeld et al., 2002), while other studies revealed an age-assessed severity of limb-muscle weakness (Doyu et al., 1992) or only a weak correlation between the decline of ADL and CAG-repeat expansion (La Spada et al., 1992). Since most of the studies performed thus far contained small sample sizes, the influence of CAG-repeat length on the clinical course of SBMA patients remains obscure. In other CAG-repeat diseases such as Huntington's disease, spinocerebellar ataxia type 3 (SCA3) and dentatorubral-pallidoluysian atrophy (DRPLA), an age-assessed residual cell population, a variety of clinical manifestations and MRI-assessed cerebellar volume have been reported to correlate with CAG-repeat length (Koide et al., 1994; Furtado et al., 1996; Penney et al., 1997; Abe et al., 1998). However, it is still not known how CAG-repeat length influences the progression and prognosis of CAG-repeat diseases.
Recent research has suggested therapeutic approaches to SBMA. In a transgenic mouse model expressing the human AR gene with expanded CAG repeats, progressive muscular atrophy and weakness associated with the nuclear accumulation of mutant AR protein was observed. These phenotypes were significantly ameliorated by anti-testosterone therapy (Katsuno et al., 2002, 2003), and the clinical and pathological phenotypes of these mice were markedly improved by the overexpression of heat shock proteins (Adachi et al., 2003; Katsuno et al., 2005). Furthermore, 17-allylamino-17-demethoxygeldanamycin (17-AAG), a potent HSP90 inhibitor, was recently shown to ameliorate motor function deficits and pathological changes in SBMA transgenic mice (Waza et al., 2005). These remarkable therapeutic effects in the transgenic mouse model strongly suggest the possibility of using these approaches in human clinical trials. In order to prepare for such a therapeutic approach, it is important to establish the natural history of clinical symptoms of SBMA based on a large number of patients.
In the present study, we investigated the natural course of SBMA as assessed by 9 ADL milestones in 223 Japanese SBMA patients, and correlated the age of onset of specific milestones during the course of the disease with the CAG-repeat length in the AR gene.
Patients and methods
Patients and clinical evaluations
Our laboratory diagnosed 303 patients as SBMA by genetic analysis between 1992 and 2004. Two-thirds of the patients were followed in Nagoya University Hospital or affiliated hospitals, while the other patients were from other hospitals nationwide. These patients were followed by neurologists from 1 year to >20 years. We reviewed the clinical course of the disease in 223 out of 303 patients. The initial symptoms and onset of nine ADL milestones were assessed to evaluate the clinical course of the disease. The ADL milestones were defined as follows: hand tremor (patient awareness of hand tremor), muscular weakness (initial patient awareness of muscular weakness in any part of the body), requirement of a handrail (patient was unable to ascend stairs without the use of a handrail), dysarthria (patient was unable to articulate properly and had intelligible speech only with repetition), dysphagia (patient choked occasionally at meals), use of a cane (patient used a cane constantly when away from home), use of a wheelchair (patient used a wheelchair when away from home) and development of pneumonia (patient developed pneumonia that required in-hospital care). The age at death and cause of death were also investigated. We assessed the age at which the ADL milestones first occurred and the age at death by direct interview, examination of the patients, family interviews and by reviewing the patient's clinical record. The milestones that could be recognized by family members, such as the use of a cane, the use of a wheelchair or the development of pneumonia, were confirmed by them wherever possible.
All evaluators used similar criteria to assess each milestone. To verify these nine ADL milestones as characteristic landmarks in the progression of SBMA symptoms, two neurologists independently assessed their onset in SBMA patients. The accordance between the evaluators of the age of onset of each ADL milestone was verified in 20 SBMA patients with Pearson's correlation coefficients ranging from 0.95 to 0.99.
The clinical landmarks adopted in the previous studies that showed clinical courses of SBMA, based on the characteristic symptoms, were onset of weakness, difficulty climbing stairs, being wheelchair-bound, tremor, gynaecomastia, fasciculations, premature exhaustion of muscles and chewing, muscle cramps, muscle pain, dysarthria and dysphagia (Doyu et al., 1992; La Spada et al., 1992; Shimada et al., 1995; Sperfeld et al., 2002; Sinnreich et al., 2004). We excluded development of gynaecomastia and fasciculation from the ADL milestones, since more than one-third of the patients were not aware of these symptoms, despite their presence. The appearance of muscle cramp and exhaustion of muscles and chewing were also excluded as their recognition was extremely variable among the patients. Some patients recognized them at a very early phase, while others did so only at later stages or not at all.
We used the modified Rankin scale (van Swieten et al., 1988) for the assessment of clinical disability in daily life and examined the serum levels of creatine kinase (CK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), total cholesterol, total testosterone and haemoglobin A1c (HbA1c) as laboratory markers for disease status. As controls, we used the serum levels of CK, AST, ALT, total cholesterol and HbA1c from health screening data of 62–70 males aged 24–79 years, free from neuromuscular diseases. For serum testosterone levels, we adopted published control data from 1143 Japanese males determined by the same assay method that we used in this study (Iwamoto et al., 2004).
We implemented the ethics guideline for human genome/gene analysis research and the ethics guideline for epidemiological studies endorsed by the Japanese government. Before we interviewed the patients, we obtained written informed consent. In cases where this was not possible, such as deceased patients, we used only existing material without informed consent and strictly protected anonymity. All of the study plans were approved by the ethics committee of Nagoya University Graduate School of Medicine.
Genetic analysis
Genomic DNA was extracted from peripheral blood of the SBMA patients using conventional techniques. PCR amplification of the CAG repeat in the AR gene was performed using a fluorescein-labelled forward primer (5′-TCCAGAATCTGTTCCAGAGCGTGC-3′) and a non-labelled reverse primer (5′-TGGCCTCGCTCAGGATGTCTTTAAG-3′). Detailed PCR conditions were described previously (Tanaka et al., 1999). Aliquots of PCR products were combined with loading dye and separated by electrophoresis with an autoread sequencer SQ-5500 (Hitachi Electronics Engineering, Tokyo, Japan). The size of the CAG repeat was analysed on Fraglys software version 2.2 (Hitachi) by comparison with co-electrophoresed PCR standards with known repeat sizes. The CAG-repeat size of the PCR standard was determined by direct sequence as described previously (Doyu et al., 1992).
Data analysis
All variables were summarized using descriptive statistics, including median, mean, SD, percentile and percentages. Age at ADL milestone data from a sufficient number of the patients was evaluated by Kaplan–Meier analyses, and log rank test statistics were used to determine whether Kaplan–Meier transition curves differed among subgroups. Relationships between the age at each ADL milestone and the length of CAG repeat of AR gene were analysed using Pearson's correlation coefficient. Correlations between laboratory test value and the age at examination were also analysed using Pearson's correlation coefficient. P-values of <0.05 were considered to be statistically significant. Calculations were performed using the statistical software package Dr SPSS II for Windows (SPSS Japan Inc., Tokyo, Japan).
Results
Clinical and genetic backgrounds of SBMA patients
A total of 223 SBMA patients were included in this study (Table 1). All of the patients were of Japanese nationality. The mean age at the time of data collection was 55.2 ± 10.5 years (range = 30–87 years). The mean duration from onset assessed by the patient's initial awareness of muscle weakness was 9.8 ± 7.2 years (range = 0–37).
Clinical and genetic backgrounds of SBMA patients
| Clinical and genetic features | Mean ± SD (range) |
|---|---|
| Age at examination (years) | 55.2 ± 10.5 (30–87) |
| Duration from onset (years)a | 9.8 ± 7.2 (0–37) |
| CAG-repeat length in AR gene (number) | 46.6 ± 3.5 (40–57) |
| Location of initial muscular weakness the patients perceived (%)b | |
| Facial | 2.4 |
| Bulbar | 11.4 |
| Upper extremities | 31.0 |
| Lower extremities | 70.5 |
| Modified Rankin scale at examination (%) | |
| 0–1 | 17.2 |
| 2–3 | 66.1 |
| 4–6 | 16.7 |
| Serum markers at examination | |
| Serum CK (n = 182) (IU/l) | 863.5 ± 762.5 (31–4955) |
| HbA1c level (n = 76) (%) | 5.7 ± 1.1 (4.3–9.6) |
| Serum testosterone level (n = 61) (ng/ml) | 6.48 ± 1.83 (2.85–10.20) |
| Serum AST (n = 130) (IU/l) | 44.3 ± 29.4 (17–238) |
| Serum ALT (n = 133) (IU/l) | 52.6 ± 37.1 (12–248) |
| Total cholesterol level (n = 82) (mg/dl) | 219.3 ± 42.3 (119–413) |
| Clinical and genetic features | Mean ± SD (range) |
|---|---|
| Age at examination (years) | 55.2 ± 10.5 (30–87) |
| Duration from onset (years)a | 9.8 ± 7.2 (0–37) |
| CAG-repeat length in AR gene (number) | 46.6 ± 3.5 (40–57) |
| Location of initial muscular weakness the patients perceived (%)b | |
| Facial | 2.4 |
| Bulbar | 11.4 |
| Upper extremities | 31.0 |
| Lower extremities | 70.5 |
| Modified Rankin scale at examination (%) | |
| 0–1 | 17.2 |
| 2–3 | 66.1 |
| 4–6 | 16.7 |
| Serum markers at examination | |
| Serum CK (n = 182) (IU/l) | 863.5 ± 762.5 (31–4955) |
| HbA1c level (n = 76) (%) | 5.7 ± 1.1 (4.3–9.6) |
| Serum testosterone level (n = 61) (ng/ml) | 6.48 ± 1.83 (2.85–10.20) |
| Serum AST (n = 130) (IU/l) | 44.3 ± 29.4 (17–238) |
| Serum ALT (n = 133) (IU/l) | 52.6 ± 37.1 (12–248) |
| Total cholesterol level (n = 82) (mg/dl) | 219.3 ± 42.3 (119–413) |
Normal values for serum CK range = 45–245 IU/l; HbA1c range = 4.3–5.8%; serum testosterone range = 2.7–10.7 ng/ml; serum AST range = 0–41 IU/l; serum ALT range = 0–45 IU/l; and total cholesterol range = 120–220 mg/dl. aOnset was assessed by patients' initial awareness of muscle weakness; bsome patients noticed muscle weakness in two locations simultaneously.
Clinical and genetic backgrounds of SBMA patients
| Clinical and genetic features | Mean ± SD (range) |
|---|---|
| Age at examination (years) | 55.2 ± 10.5 (30–87) |
| Duration from onset (years)a | 9.8 ± 7.2 (0–37) |
| CAG-repeat length in AR gene (number) | 46.6 ± 3.5 (40–57) |
| Location of initial muscular weakness the patients perceived (%)b | |
| Facial | 2.4 |
| Bulbar | 11.4 |
| Upper extremities | 31.0 |
| Lower extremities | 70.5 |
| Modified Rankin scale at examination (%) | |
| 0–1 | 17.2 |
| 2–3 | 66.1 |
| 4–6 | 16.7 |
| Serum markers at examination | |
| Serum CK (n = 182) (IU/l) | 863.5 ± 762.5 (31–4955) |
| HbA1c level (n = 76) (%) | 5.7 ± 1.1 (4.3–9.6) |
| Serum testosterone level (n = 61) (ng/ml) | 6.48 ± 1.83 (2.85–10.20) |
| Serum AST (n = 130) (IU/l) | 44.3 ± 29.4 (17–238) |
| Serum ALT (n = 133) (IU/l) | 52.6 ± 37.1 (12–248) |
| Total cholesterol level (n = 82) (mg/dl) | 219.3 ± 42.3 (119–413) |
| Clinical and genetic features | Mean ± SD (range) |
|---|---|
| Age at examination (years) | 55.2 ± 10.5 (30–87) |
| Duration from onset (years)a | 9.8 ± 7.2 (0–37) |
| CAG-repeat length in AR gene (number) | 46.6 ± 3.5 (40–57) |
| Location of initial muscular weakness the patients perceived (%)b | |
| Facial | 2.4 |
| Bulbar | 11.4 |
| Upper extremities | 31.0 |
| Lower extremities | 70.5 |
| Modified Rankin scale at examination (%) | |
| 0–1 | 17.2 |
| 2–3 | 66.1 |
| 4–6 | 16.7 |
| Serum markers at examination | |
| Serum CK (n = 182) (IU/l) | 863.5 ± 762.5 (31–4955) |
| HbA1c level (n = 76) (%) | 5.7 ± 1.1 (4.3–9.6) |
| Serum testosterone level (n = 61) (ng/ml) | 6.48 ± 1.83 (2.85–10.20) |
| Serum AST (n = 130) (IU/l) | 44.3 ± 29.4 (17–238) |
| Serum ALT (n = 133) (IU/l) | 52.6 ± 37.1 (12–248) |
| Total cholesterol level (n = 82) (mg/dl) | 219.3 ± 42.3 (119–413) |
Normal values for serum CK range = 45–245 IU/l; HbA1c range = 4.3–5.8%; serum testosterone range = 2.7–10.7 ng/ml; serum AST range = 0–41 IU/l; serum ALT range = 0–45 IU/l; and total cholesterol range = 120–220 mg/dl. aOnset was assessed by patients' initial awareness of muscle weakness; bsome patients noticed muscle weakness in two locations simultaneously.
The mean number of CAG repeats in the AR gene was 46.6 ± 3.5 (range = 40–57). The location of the initial noticeable muscular weakness was lower extremities in 70.5%, upper extremities in 31.0%, bulbar symptoms in 11.4% and facial weakness in 2.4%. Some patients noticed muscle weakness initially in two locations simultaneously; thus overlap between the groups existed. Weakness in the lower extremities was noticed most often as difficulty in climbing stairs, followed by difficulty in walking for long distances and difficulty in standing from a sitting position. Bulbar symptoms were first noticed as a difficulty in articulating properly. ADL assessed by a modified Rankin scale at examination was 0–1 in 17.2%, 2–3 in 66.1% and 4–6 in 16.7% of the patients. Serum CK levels were 863.5 ± 762.5 IU/l (range = 31–4955; normal value = 45–245 IU/l), HbA1c levels were 5.7 ± 1.1% (range = 4.3–9.6; normal value = 4.3–5.8%), serum testosterone levels were 6.48 ± 1.83 ng/ml (range = 2.85–10.20; normal value = 2.7–10.7 ng/ml), serum AST levels were 44.3 ± 29.4 IU/l (range = 17–238; normal value = 0–41 IU/l),serum ALT levels were 52.6 ± 37.1 IU/l (range = 12–248; normal value = 0–45 IU/l) and serum total cholesterol levels were 219.3 ± 42.3 mg/dl (range = 119–413; normal value = 120–220 mg/dl).
Age at which ADL milestones appear
Age distributions at which the ADL milestones initially appeared are summarized in Fig. 1. Hand tremor was the earliest of the ADL milestones that the patients noticed, and it occurred at a median age of 33 years. Hand tremor was particularly noticed when patients used their hands such as in holding a drinking glass. Muscular weakness, predominantly in the lower extremities, was noticed at a median age of 44 years, followed by the need of a handrail when going up stairs at a median age of 49 years. Dysarthria, dysphagia and the use of a cane appeared at median ages of 50, 54 and 59 years, respectively. The use of a wheelchair started at a median age of 61 years. Patients developed pneumonia owing to aspiration and required in-hospital care at a median age of 62 years. The median age of those 15 patients who died before this report was 65 years. The predominant cause of death in eight of these cases was aspiration pneumonia. One patient died of lung cancer, and another patient died from ischaemic heart disease. One patient committed suicide. The causes of death of the other four patients were unknown. The ages of onset of each ADL milestone showed a considerable wide-ranged distribution from 25 to 30 years when assessed from the 10th to 90th percentile range. Although there were no significant differences in the mean number of CAG repeats in the AR gene of the patients in which we assessed the age at onset of each ADL milestone (Fig. 1), suggesting that age distributions at each milestone were derived from genetically uniform patients, we tested the hypothesis that the wide range in ages of onset at each milestone may be due to individual differences in CAG-repeat lengths.
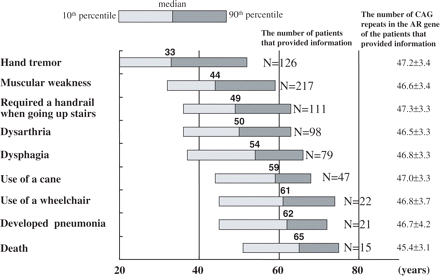
Age distribution of ADL milestones for 223 SBMA patients. The mean number of CAG repeats in the AR gene of the patients does not differ significantly as shown at the right.
Age at onset of each ADL milestone correlates well with CAG-repeat length
As shown in Fig. 2, the onset age of the individual ADL milestones examined showed significant correlations with the CAG-repeat length of the patients reporting on these symptoms (r = −0.853 to −0.447, P < 0.016–0.001). Of these, age at onset of hand tremor, requirement of a handrail, use of a wheelchair, developing pneumonia requiring in-hospital care and death were strongly correlated with the CAG repeats with r < −0.5. Furthermore, the onset ages of pneumonia and death were highly correlated with the CAG repeats with r = −0.78 and −0.85, respectively, indicating that these specific events, the onset ages of which the patients or their families were able to indicate more definitely, showed a more significant correlation with the CAG-repeat length than other ADL milestones.
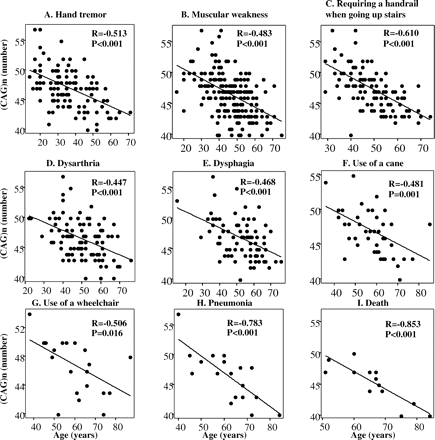
(A–I) Correlation of the CAG-repeat number of and the age at each ADL milestone. There were significant correlations between CAG number and age at all milestones analysed using Pearson's correlation coefficient.
Since 47 repeats was the median CAG-repeat length of the entire patient group, we further compared the Kaplan–Meier curves for age at onset of hand tremor, muscular weakness and requirement of a handrail between the patient group with 47 CAG repeats or more and those with <47 CAG repeats (Fig. 3). We assessed only these three ADL milestones, since the number of patients in these groups was sufficient to perform a log rank test analysis. The patients with <47 CAG repeats showed regression curves shifted by ∼10 years compared with those with ≥47 CAG repeats (Fig. 3, P < 0.001 in log rank test). Together, these observations strongly suggest that the onset age of each ADL milestone is highly dependent on CAG-repeat length in the AR gene.
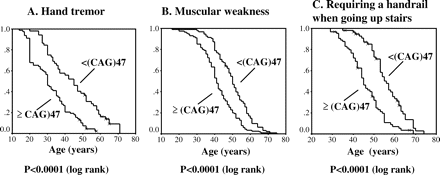
(A–C) Kaplan–Meier analysis of age at onset of hand tremor, muscular weakness and requirement of a handrail. There was a highly significant difference between the patient group with ≥47 CAG repeats and the group with <47 CAG repeats, as compared by log rank tests.
CAG-repeat length does not correlate with the rate of disease progression assessed by ADL milestones
In order to assess whether CAG-repeat length influences the disease progression rate, we examined the relationship between the time intervals from onset age of muscular weakness to that of requirement of a handrail when going up stairs, use of a cane, use of a wheelchair, development of pneumonia and death and the CAG-repeat lengths in these groups (Fig. 4). We did not find any significant correlations of the intervals among the onset age of the various milestones with the CAG-repeat length, suggesting that the progression rate of the disease is not significantly influenced by the CAG-repeat size.
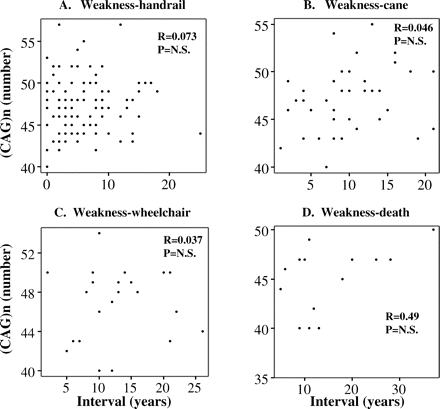
(A–D) Correlation between the AR gene CAG number and the time interval between the ADL milestones. The time interval from the age at first awareness of muscular weakness to the age at requirement of a handrail, use of a cane, use of a wheelchair and death were compared with the CAG number by Pearson's correlation coefficient. There were no significant correlations in any of the interval times.
In addition, we examined the declining regression assessed by those ADL milestones in individual patients with ≥47 CAG repeats compared with those with <47 (Fig. 5). These regression lines were divergent from each other, possibly because of divergent CAG-repeat size, while the mean slopes of the regression lines of the two groups were likely to be parallel. There were no significant differences between the interval times of the two groups as assessed by unpaired t-test (Fig. 5), suggesting, again, that the rate of disease progression was not markedly dependent upon the size of the CAG repeats.
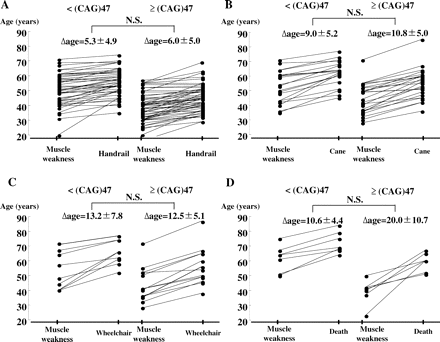
(A–D) Individual case presentation of the declining regression assessed by ADL milestones. The interval times from the age at first awareness of weakness to the age at requirement of a handrail, use of a cane, use of a wheelchair and death are described for individual patients from two groups, those with <47 CAG repeats and those with ≥47 repeats. These regression lines were divergent from each other, possibly owing to divergent CAG-repeat size, while the mean slopes of the regression lines were likely to be parallel among the two subgroups of patients. There were no significant differences between the interval times of the two groups of patients analysed by unpaired t-test.
Age-related changes of laboratory data and their relation to CAG repeats
Glucose intolerance, serum CK and ALT elevation and androgen insensitivity of SBMA patients have been reported (Sobue et al., 1989; Shimada et al., 1995; Dejager et al., 2002; Sinnreich et al., 2004). We examined the relationship between serum CK, HbA1c, testosterone, total cholesterol, AST and ALT levels and the age and CAG-repeat length of the patients. The serum levels of CK, AST and ALT were elevated in subpopulations of patients, particularly in the early phase of the disease, while these levels gradually declined with age (Fig. 6A, E and F). In advanced ages, the levels of these serum markers had declined to nearly normal levels. Serum testosterone levels were slightly elevated from control values in one-third of patients; in general, they declined slightly with age (Fig. 6C). However, even at these advanced ages testosterone levels were within or above the normal range. In contrast, HbA1c levels were within the normal range in the patients with short disease durations, but they gradually increased to above the normal range as the age of patients increased (Fig. 6B). Cholesterol levels were mildly elevated in some patients, but there was no particular age-dependent change observed (Fig. 6D). Elevated levels of these serum markers were not correlated with the CAG-repeat sizes (data not shown). Therefore, the levels of these markers appear to reflect the active pathological process of the disease, especially in the early or late phases, but their significance should be examined further.
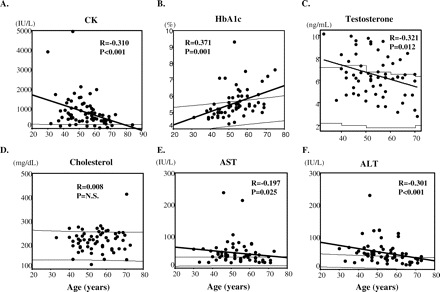
(A–F) Correlation between the levels of serum markers and the age at examination. A weak, but significant, correlation was seen between HbA1c and age, while weak, but significant, inverse correlations were seen between CK, testosterone, AST and ALT and age as analysed by Pearson's correlation coefficient. Cholesterol levels were not correlated with age. The thin lines in each plot indicate the 95% confidence intervals calculated from control subjects.
Discussion
Our study elucidated the natural history of SBMA patients based on nine ADL milestones. SBMA progressed slowly to the end stage with a median duration from onset assessed by muscle weakness to the appearance of pneumonia of 16 years, and to death of 22 years whereas the median durations from age of onset to the age of requirement of a handrail, dysarthria and dysphagia were 5, 6 and 10 years, respectively, indicating that the ADL deterioration leading to a decline in the quality of daily living during early phases of the diseases is significant, in spite of a relatively long lifespan. The lifespan of SBMA patients was previously speculated to be 10–15 years shorter than those of the general Japanese male population (Mukai, 1989). In this study, 15 of the 223 patients died at a median age of 65 years. Although there are too few data to make a reliable calculation, this is ∼12 years shorter than that of the current lifespan of the normal Japanese male indicated by the abridged life table announced by the Japanese Ministry of Health, Labor and Welfare in 2003, and, thus, is consistent with the previous speculation (Mukai, 1989). Of these 15 patients, the most common cause of death was pneumonia due to aspiration and dysphagia. Thus, the bulbar symptoms, such as difficulty in proper articulation and mild dysphagia, were relatively mild in their early manifestations, but were serious symptoms in the late phase of the disease, when the patients were prone to death. The progression was apparently slower than that of ALS, another adult-onset motoneuron disease, which occasionally mimics SBMA phenotypes, particularly in the early phase (La Spada et al., 1992; Parboosingh et al., 1997; Traynor et al., 2000).
The onset ages of each ADL milestone were extremely variable, but all were well correlated with the CAG-repeat size in the AR gene. Patients with longer CAG-repeat sizes showed an earlier onset age of each ADL milestone examined, including occurrence of pneumonia or death in the end stage. Several previous studies also documented the natural history of SBMA. They showed that the age of disease onset assessed by muscle weakness was strongly correlated with AR gene CAG-repeat size (Doyu et al., 1992; Igarashi et al., 1992; La Spada et al., 1992; Shimada et al., 1995), whereas the onset ages of other symptoms such as fatigue, tremor, occurrence of gynaecomastia and severity of muscle weakness were not significantly correlated with repeat size (La Spada et al., 1992; Amato et al., 1993; Mariotti et al., 2000; Dejager et al., 2002; Sperfeld et al., 2002). It is not clear why the relations between the onset age of these symptoms and CAG repeat size were not apparent in these reports, since significant correlations with the onset age of hand tremor and muscular weakness were confirmed in the present study. One possibility may be the relatively small sample sizes in the previous studies (Amato et al., 1993; Sperfeld et al., 2002). An alternative explanation may be that very early symptoms, such as gynaecomastia and fatigue, are not very accurate or reliable markers for ADL milestones compared with later symptoms, especially in a retrospective study. Indeed, the correlation of the onset age of tremor with CAG-repeat size was weaker than that of the onset age of the use of a wheelchair or a cane, which were more advanced ADL milestones used in our study.
The relationship between CAG-repeat size, disease markers and rate of disease progression have also been assessed extensively in Huntington's disease (Illarioshkin et al., 1994; Brandt et al., 1996; Furtado et al., 1996; Penney et al., 1997; Rosenblatt et al., 2003). Neuronal loss in the caudate nucleus and putamen, adjusted for age of death, correlated well with CAG-repeat length (Furtado et al., 1996; Penney et al., 1997; Rosenblatt et al., 2003). The rate of progression assessed by symptom severity controlled by duration from onset (Illarioshkin et al., 1994) also correlated strongly with the CAG-repeat size. In addition, we previously demonstrated that the extent of cerebellar atrophy and severity of muscle weakness, both adjusted by age at examination correlated well with CAG-repeat size in SCA3 and SBMA, respectively (Doyu et al., 1992; Abe et al., 1998). These observations suggested that longer CAG repeats resulted in an earlier age at onset and greater neuronal loss when compared with shorter repeats. There is also some evidence that they contribute to a faster rate of clinical decline.
In our present study, as documented in Figs 2 and 3, patients with longer CAG repeats reached each of the ADL milestones such as hand tremor, muscle weakness or the requirement of a handrail when going up stairs much earlier than did the patients with shorter CAG repeats. Interestingly, however, the decline curves, as documented with Kaplan–Meier analyses, for these individual milestones were similar (Fig. 3), with an ∼10-year difference between the patients with ≥47 repeats and those with <47 repeats. The earlier age at onset for each ADL milestone in patients with longer repeat lengths is similar to observations in cases of Huntington's disease.
The most striking observation in our study was that the interval periods between individual ADL milestones, such as between onset of muscle weakness and that of requirement of a handrail, use of a cane, being wheelchair-bound or death were not affected by the CAG-repeat length (Figs 4 and 5). Although patients with longer CAG-repeat size reached individual ADL milestones faster than those with shorter repeats, the decline rate from one ADL milestone to another was not influenced by the CAG-repeat size. These results suggest that the rate of disease progression assessed by ADL milestones is not influenced by CAG-repeat length.
Therefore, we may propose a view simulating the natural history of SBMA, in that, the decline curves of ADL in the SBMA patients with longer CAG-repeat size are shifted earlier than those in the SBMA patients with shorter CAG-repeat size, and the slopes of the decline curves are parallel to one another.
The phenotypic decline curves provided by mouse models of CAG-repeat diseases (Adachi et al., 2003; von Horsten et al., 2003) also support our view of the natural history of SBMA. The present findings are informative in understanding the pathophysiology of SBMA. CAG-repeat size is known to be a determinant factor for the entry of neuronal cells harbouring a mutant AR gene with expanded CAG repeats into the neuronal degeneration process in vitro, as well as in vivo (Mangiarini et al., 1996). However, it is not known whether the rate of neuronal degeneration leading to subsequent cell death is dependent on CAG-repeat size. Once neuronal cell degeneration or neuronal cell dysfunction is initiated, the progression of degeneration to cell death may be determined by intrinsic factors such as a cell death processing system other than the CAG-repeat size. Thus, we suggest that the onset time of certain ADL milestones reflects how many neurons have entered into the neurodegeneration-dysfunction process, which is determined by CAG-repeat size, rather than the intrinsic cell death process.
Recently, we demonstrated that several interventions, anti-testosterone therapy with leuprorelin (Katsuno et al., 2003), induction of Hsp70 (Adachi et al., 2003; Katsuno et al., 2005), inhibition of HDAC (Minamiyama et al., 2004) or inhibition of Hsp90 (Waza et al., 2005) showed potent therapeutic effects in improving the characteristic phenotypes and pathology in the SBMA transgenic mouse model. These observations strongly encourage the application of the therapeutics to human SBMA patients. Unlike the therapeutic approach commonly taken in neurodegenerative diseases of replacing lost substances such as neurotransmitters, these new therapeutics ameliorate the disease progression itself by preventing pathological molecular processes. Since the progression of SBMA is slow, clinical end-points will be useful for efficiently assessing the effectiveness of these therapies. The present study may indicate ADL milestones that can be clinical end-points in therapeutic trials. However, assessing the ADL milestones adopted in this study, such as the use of a cane or a wheelchair, would take years during clinical trials. Thus, we need to find a shorter-term surrogate marker that reflects the pathological process, although a genuine clinical therapeutic end-point should be examined to determine whether the ADL milestones are effectively delayed by the therapeutic intervention.
One interesting observation in this study is that serum testosterone levels were maintained at relatively high levels, even at advanced ages, although they did decrease with age (Fig. 6C). Since testosterone is an important triggering factor for polyglutamine-mediated motoneuron degeneration (Katsuno et al., 2002, 2003), these findings suggest that anti-testosterone therapy with leuprorelin (Katsuno et al., 2003) may be applicable even in aged SBMA patients.
The advantages of our study over previous studies are the large sample size and the employment of marked and apparent ADL milestones that the patients recognized easily. Nevertheless, several limitations are also present. One major limitation is that the study was retrospective in design and the decline curve was not successively and prospectively assessed in individual patients. A prospective study that follows individual patients in assessing the ADL milestones is needed to ascertain the validity of this natural history of SBMA.
The ADL milestones that we adopted for this study were selected with the assumption that they could be accurately assessed by us, the patients or family members, even in a retrospective study. However, as we demonstrated, the development of pneumonia and death showed higher significant correlations with CAG-repeat size than did other earlier ADL milestones such as the appearance of hand tremor or dysarthria, suggesting that these critical end-stage events may be more accurately assessed in a retrospective manner. We need further long-standing prospective studies to assess the disease progression more properly.
This study was supported by a COE grant from the Ministry of Education, Science and Sports of Japan and grants from the Ministry of Welfare, Health and Labor of Japan.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}